

Ijraset Journal For Research in Applied Science and Engineering Technology
Newly Synthesized Reagent [TREN 3 (2 Quinoline)] for Determination of Nickel (II) in Biological Samples by Extractive-Differential Pulse Polarography
Authors: S. Hemanth Phani Kumar, Dasari Rekha
DOI Link: https://doi.org/10.22214/ijraset.2024.59356
Certificate: View Certificate
Abstract
The synthesis and application of a novel analytical reagent, [TREN 3 (2 Quinoline)], was detailed for the precise determination of nickel (II) concentrations in biological samples and plant materials. The analysis involved extracting the nickel(II)-TREN3(2Quinoline) complex from chloroform before injection into the instrument. To ensure accuracy and various parameters such as pH, pulse amplitude, scan rate, and choice of solvent were meticulously optimized. A calibration curve constructed in the concentration range from 0.05 to 42 µg mL-1 under pH 4.0, conditions using an acetate buffer, exhibiting a high correlation coefficient of 0.992. The study also thoroughly investigated potential interference from diverse ions, Validation of the proposed method was conducted using Certified Reference Materials provided by the National Institute of Standard Technology, affirming its accuracy and reliability. Furthermore, the reagent underwent characterization through techniques including infrared spectroscopy (IR), mass spectrometry (Mass), and elemental analysis, bolstering confidence in its suitability for analytical purposes.
Introduction
I. INTRODUCTION
Nickel(II) exposure exceeding 0.05 mg/kg in the environment poses significant toxicological risks to both human beings and animals, as noted in previous studies [1,2]. Despite its widespread applications, including batteries, welding rods and wires, electronic equipment, pigments for paints and ceramics, consumer products, steel, and magnets. Nickel (II) also presents potential health hazards and can contribute to various diseases. Given these toxicological concerns, monitoring nickel (II) levels becomes paramount for safeguarding human health and environmental integrity. Developing analytical methods capable of detecting trace levels of nickel (II) is highly desirable, enabling proactive assessment and management of potential risks posed by its presence in various settings. Various analytical techniques have been proposed for monitoring of nickel(II) levels in environmental matrices, including spectrophotometry(3,4), X-ray fluorescence spectrophotometry(5) atomic absorption spectrometry(6) inductively coupled plasma atomic emission spectrometry(7,8), inductively coupled plasma mass spectrometry(9)voltammetry(10.11), and chromatography(12). However, many of these methods are either costly, prone to interference, or require specialized expertise. Consequently, despite being an older technique, the simplicity and effectiveness of the differential pulse polarography (DPP) method make it a viable option for trace analysis.
Given the typically low concentrations of nickel in environmental samples, solvent extractive and differential pulse polarographic methods prove to be suitable for overcoming this challenge. Therefore, a simple and cost-effective differential pulse polarography method was developed after extracting the metal complex from chloroform in biological samples and plant materials. The formation of the metal complex occurred through the interaction of nickel(II) with a newly synthesized analytical reagent, TREN 3 (2 Quinoline), under ordinary laboratory conditions. Various parameters were evaluated to optimize the method. This developed approach enables efficient determination of nickel(II) at parts per million (ppm) levels and demonstrates minimal interference. The method was successfully applied to determine nickel(II) concentrations in biological and plant materials. Comparison of the obtained nickel analysis results with those from established methods and certified values further validates the effectiveness and reliability of the proposed method.
II. EXPERIMENTAL
A. Apparatus
The experimental setup for the differential pulse polarography (DPP) measurements involved an Elico CL-362 model polarographic analyzer equipped with a three-electrode system. The working electrode utilized a Hanging mercury drop electrode, while an Ag/AgCl/KCl electrode served as the reference electrode, and a platinum wire acted as the auxiliary electrode. The entire system was complemented with an LX 300+ X-Y recorder to capture data. For cyclic voltammetric measurements, a Metrohm 757 VA Computrace Model AFRDE 4 potentiostat was employed in conjunction with an MSRX speed control unit supplied by Pine Instrument Company (USA), coupled with a digital electronics 200X-y/t recorder. pH values were measured using an Elico Li-129 Model glass-calomel combined electrode, sourced from Elico Ltd, Chennai, India. All experiments were conducted at a controlled temperature of 25±1°C to ensure consistency and reproducibility of results.
B. Reagents
In the experimental procedures, analytical reagent-grade chemicals and double-distilled water were exclusively utilized. TREN3(2 Quinoline), organic solvents, as well as acids including HCl, HNO3, H2SO4, and Perchloric acid, along with acetic acid and sodium acetate, were procured from Merck Chemicals. For the preparation of a 0.01 M stock solution of nickel(II), 2.377g of NiCl2•6H2O was dissolved in double-distilled water within a 100 mL volumetric flask. A freshly prepared working standard solution was derived by diluting the stock solution with double-distilled water just prior to analysis. Similarly, a 0.001 M stock solution of TREN3(2 Quinoline) was prepared by dissolving an appropriate quantity of the reagent in 100 mL of methanol. The working solution was then prepared through suitable dilution with the same solvent. Acetate buffer solutions with pH values spanning from 3 to 11 were prepared by mixing 0.1 M acetic acid and 0.1 M sodium acetate in appropriate ratios, with a concentration of 13.6 g/L. This meticulous approach to chemical selection and solution preparation ensures the reliability and accuracy of the experimental results.
C. Synthesis of TREN3(2 Quinoline)]
An Equimolar ratio of Tris(2-aminoethyl)amine (Tren) (1.0 g) and Quinoline (1.0 g) in methanol mixture was refluxed for 3 to 4 hrs and the contents were cooled at room temperature creating an orange-red colour precipitate. The resulting precipitate was filtered (Whatman filter paper No. 44) and washed with 50 mL ethanol. The crude solid was recrystallized from aqueous ethanol and dried on CaCl2, with a Yield=97 % and M.P-115-120 oC. Infrared absorption spectrum was shown in (Figure 1).
D. General Procedure for the Determination of Nickel(II)
In the experimental procedure, an aliquot of the working standard solution (0.001 M) containing a variable volume ranging from 1 to 100 µL (equivalent to 0.2377 g of metal ion) was dispensed into a 25 mL volumetric flask. To this, 5 mL of acetate buffer solution with a pH of 4.0 was added, followed by the addition of 2 mL of the reagent solution, which is soluble in methanol. The resulting mixture was vigorously shaken with 5 mL portions of chloroform for 30 seconds and allowed to stand for 5-10 minutes to facilitate phase separation. Subsequently, the organic layer was carefully collected and transferred into a polarographic cell, followed by dilution with 9 mL of acetate buffer solution. The cell contents were then deoxygenated with nitrogen gas for 10 minutes to minimize interference from oxygen. After recording the initial polarogram, small increments (0.2 mL) of the standard solution were sequentially added to the cell, with each addition followed by one minute of treatment, and subsequent polarograms were recorded under similar conditions. This process was repeated for a total of 10 additional times. Notably, the highest precision was achieved under conditions of pH 4.0, exhibiting a correlation factor of 0.992 and a relative standard deviation (RSD) of 0.7%. The optimal parameters for the polarographic measurements included a drop time of 2 seconds, a pulse amplitude of 50 mV, a scan rate of 12 mV/s, and an applied potential of -613.0 mV. These meticulously controlled experimental conditions ensure the accuracy and reproducibility of the obtained results.
E. Sample Collection
The study area selected for this research is Tirupati, a small town located in the southern region of India. Despite its relatively modest size, Tirupati holds global recognition due to its rapid growth and significance as a pilgrimage destination. The primary attraction drawing thousands of visitors daily is the Lord Balaji Temple (Tirumala), with an estimated influx of 50,000 to 60,000 pilgrims each day. However, this continuous flow of visitors poses a threat to the natural beauty of Tirupati. Additionally, the town hosts several small and medium-scale industries situated in its suburban fringes. Due to these factors, Tirupati, known for its religious significance, has been chosen as the ocal area for sample collection in this study.
Biological and plant samples were gathered from various locations within and around Tirupati to represent the diversity of the study area. Throughout the sampling process, meticulous precautions were taken to ensure the integrity of the samples. This included careful selection of sample containers, proper collection and storage procedures, as well as rigorous processing and analysis protocols. By adhering to these measures, the research aims to provide a comprehensive understanding of the environmental conditions in Tirupati
F. Analysis of Biological Samples
In the process of preparing hair samples for analysis, several steps were undertaken. Initially, the hair samples were washing with acetone, repeated 2 to 3 times in a beaker with continuous stirring to remove any external contaminants. Following this, the washed samples were dried in an electric oven at 70°C for a duration of 4 hours to ensure complete removal of moisture. Subsequently, 2.0 grams of the dried sample were weighed and transferred into a beaker. To the weighed sample, a mixture of nitric acid and perchloric acid in a 1:1 ratio was added. The resulting mixture was heated on a hot plate until it reached near dryness, facilitating the dissolution of the sample and decomposition of organic matter. Once evaporated to near dryness, the ash residue was dissolved in 5 mL of hydrochloric acid (1+9) and further evaporated to dryness. The resulting residue was dissolved in 2 mL of hydrochloric acid (35.5%) and then filtered to remove any insoluble particles. The filtrate was then diluted to a final volume of 25 mL with water. From this prepared solution, appropriate volumes were taken for the determination of nickel(II) using the previously described general procedure. The obtained results were tabulated and presented in Table 2, providing insights into the nickel(II) content in the analyzed hair samples.
G. Analysis of Plant Material
The process for preparing the freshly collected Pisum sativum (Hulls), Mangifera indica, Eucalyptus, and Azadirachta indica leaves samples were involved in several steps. Firstly, 5.0 grams of each sample were placed in a 250 mL beaker. Subsequently, a solution comprising concentrated H2SO4/HNO3 in a 1:1 (v/v) ratio (10 mL) was added to the beaker. The mixture was then heated until the solution became clear. Following this, the solution was filtered using Whatman filter paper No. 44 to remove any insoluble particles. The filtrate was then concentrated in a porcelain bowl until its volume reached approximately 5 mL. After cooling, the solution was diluted up to a final volume of 50 mL using deionized double-distilled water. Once the solution was prepared, 1 mL of it was subjected to the general procedure described earlier for the determination of nickel(II). The obtained results were tabulated and presented in Table 3, Furthermore, detailed examination of plant tissues, specifically Umbilicaria muhlenbergii, was conducted following a reference procedure outlined in a previous study [13]. The results obtained from this examination were then compared with the reported method, and the comparative findings were presented in Table 4. The accuracy and precision of the present method were validated by analyzing the Certified Reference Materials (CRM’s) which was distributed by the National Institute of Standard Technology (NIST) of Rice Flour (NIES-CRM-10A), Wheat Flour (ARC/CL-WF), and Rye Bread Flour (CSRM-12-2-05). Inter calibration was performed by using above said materials and the analytical results obtained from the present method strongly agreed with the Certified Reference Materials (CRM’s) and the data were mentioned in Table 5.
III. RESULTS AND DISCUSSION
A. Differential Pulse Polarographic Studies
- Effect of pH, Choice of Solvent and Scan Rate
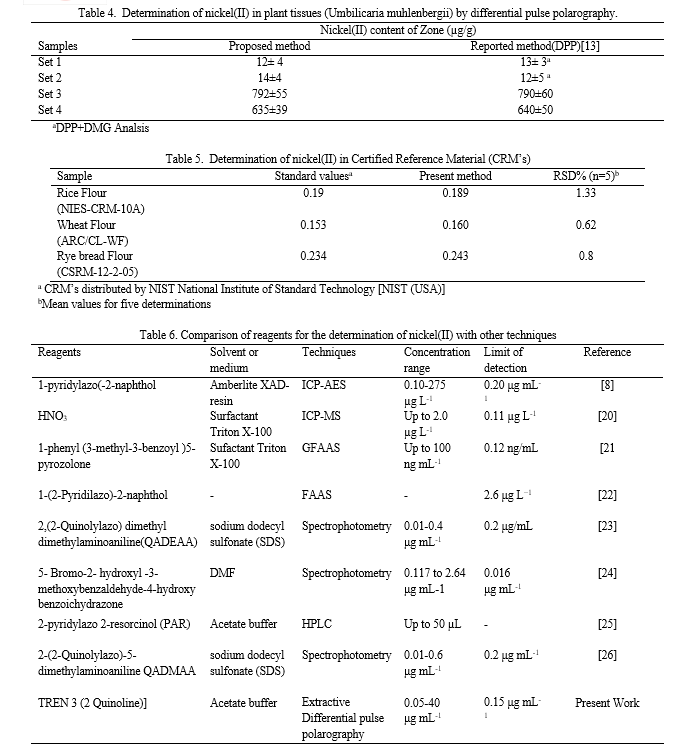
The influence of pH on the determination of nickel(II) was explored through differential pulse polarography, focusing on the peak potential (Ep) and current intensity (ip) of the [Ni TREN 3 (2 Quinoline)] complex extracted from chloroform. pH variations ranging from 2.0 to 10.0 were examined, revealing that the maximum peak current was achieved at pH 4.0, corresponding to a peak potential of -615.0 mV, as depicted in Figure 2. Notably, as the pH was increased from 2.0 to 10.0 while maintaining a constant potential, a decrease in the formation of the metal complex was observed. Consequently, an acetate buffer of pH 4.0 was optimal for the determination of nickel(II), offering enhanced sensitivity and selectivity for subsequent investigations. Furthermore, the extraction of the nickel(II)-[TREN3(2 Quinoline)] complex was evaluated across various organic solvents, including toluene, benzene, n-butanol, dimethyl formaldehyde, carbon tetrachloride, cyclohexane, and chloroform. Among these solvents, chloroform emerged as the most suitable due to its superior extraction efficiency and maximum peak current, attributed to its polarity. Additionally, the impact of scan rate on the electrochemical response was investigated over a range of 4 to 18 mV/s. It was observed that the peak current increased with rising scan rates; however, beyond 12 mV/s, a decrease in peak current was noted. Therefore, a scan rate of 12 mV/s was selected for further studies, striking a balance between peak current enhancement and maintaining stability in the electrochemical measurements.
2. Effect of Foreign Ions
The selectivity of the proposed method was significantly enhanced through a comprehensive study of diverse ions, aimed at determining nickel(II) concentrations in biological samples and plant materials. Table 1 presents the results obtained from these investigations. In this study, foreign ions were introduced into a solution containing 10 µg/mL of nickel(II), and measurements were conducted under optimized conditions. Many of the diverse ions were found to be tolerable up to their maximum levels by utilizing appropriate masking agents, with errors remaining within acceptable limits (>2%) during the analysis of nickel(II). However, for certain ions, such as chromium, sufficient amounts of EDTA solution were added to effectively mask their interference. Additionally, the presence of low concentrations of As3+ at 40°C was employed to mitigate the interference of chromium ions. Interference from ions such as Ca2+ and Sr2+ was eliminated through prior extraction with KI/isobutyl methyl ketone. Furthermore, ions such as sodium, potassium, chlorides, nitrites, sulfates, bicarbonates, phosphates, and others did not interfere with the determination of trace metals up to concentrations of 50 µg/mL. Consequently, nearly all results were quantified in the presence of diverse ions, thereby enhancing the feasibility and selectivity of the proposed method. These findings underscore the robustness of the developed method in accurately determining nickel(II) concentrations in complex sample matrices, facilitating its applicability in practical analytical scenarios.
Top of Form
3. Calibration, Detection Limit and Precision
The calibration curve was generated using the general procedure under optimized conditions, in a concentration range of 0.05 to 42 µg/mL, yielding a high correlation coefficient of 0.992. Additionally, the detection limit of the method was determined to be 0.15 µg/mL of the final solution, obtained from five individual replicates of nickel(II) solution with a relative standard deviation of 0.7%. The relative typical calibration curve, depicted in Figure 3, was constructed based on the standard deviation (S.D.) values. Moreover, the Method of Quantification (MOQ) value was calculated utilizing the calibration curve obtained from the analytical procedure. Specifically, the MOQ for nickel(II) in biological samples was determined to be 7.940 µg/mL, providing a benchmark for the quantitative analysis of nickel(II) in complex sample matrices. These parameters collectively shows the reliability, sensitivity, and efficacy of the proposed method for the determination of nickel(II) concentrations, thereby facilitating its practical application in various analytical settings.
The proposed method was successfully implemented for the determination of nickel(II) in biological samples and plant materials, with the results presented in Tables 1, 2, 3 and 4. The validity of the method was further confirmed by analyzing Certified Reference Materials (CRMs), the results of which are shown in Table 5. Additionally, a comparison was made with several other analytical reagents synthesized for the determination of nickel(II) in various environmental samples, as presented in Table 6. This comparative analysis highlights the sensitivity of the proposed method across different solvent media, techniques employed for nickel(II) determination, concentration ranges, and detection limits. By leveraging a simple chloroform extraction procedure prior to differential pulse polarography (DPP), the proposed method demonstrates the effectiveness in accurately quantifying nickel(II) in diverse sample matrices. The comparison with other analytical reagents further underscores the advantages and unique capabilities of the proposed method, positioning it as a valuable tool for nickel(II) analysis in environmental and biological samples.

Conclusion
A novel and environmentally friendly method for the determination of nickel (II) in biological samples and plant materials was developed, featuring simplicity, sensitivity, and rapidity. This method incorporates a preconcentration step following a chloroform extraction procedure. Key to this method is the synthesis of a new analytical reagent, TREN 3(2 Quinoline), which forms stable complexes with nickel(II). The presence of the selective functional group -C=N- in the complex facilitates a robust response, easily reducible at the electrode surface of the polarograph. Additionally, this reagent effectively suppresses interference from diverse ions, including cations, anions, and other salts, through the strategic use of masking agents. Overall, this innovative approach offers a reliable and efficient means of analyzing nickel(II) in complex sample matrices, contributing to advancements in environmental and biological analysis methodologies.
References
[1] J.J. Scott-Fordsmand, P.H. Krogh and S.P. Hopkin, Ecotoxicology Environmental Safty, 43(1), 57 (1999). [2] K.S. Kasprzak. Mutation research, 533(1-2), 67(2003),. [3] E.Y. Hashem, M.S. Abu-Bakr, S.M. Hussain, Spectrochemica Acta Part A, 59, 761(2003). [4] Y. Liu, X. Chang. S. Wang.Y. Guo, B. Din, S. Meng, Talanta, 64 (1), 160(2004). [5] Z. Augustynski, B. Dziunikowski, J. Meitin, Journal of Radioanalytical Nuclear Chemistry, 63 (2), 325 (1981). [6] J.L.Manzoori, A. Bavili-Tabrizi, Microchimca Acta, 141(3-4), 201(2003). [7] E. Vereda Alonso, A. Garcia de Torres, J.M. Cano Pavon, Mikrochimca Acta, 110(1-3), 41(1993). [8] S.LC. Ferreira, C.F. de Brito, A.F. Dantas. N.M. Lopo de Araujo, A.C.S. Costa, Talanta, 48(5), 1173(1999). [9] C.J. Park, S.A. Yim, Journal of Analytical Atomic Spectrom, 14, 1061(1999). [10] M. Korolczuk, Talanta, 53(3), 679(2000). [11] N.Y. Sreedhar, D. Rekhs, Analytical chemistry An Indian journal, 8(2), 209(2009). [12] X. Yang. Z. Qiao. W. Wei, S.Yao, Talanta, 46(4), 697(1998). [13] C.J. Flora, E. Nieboer. Analytical chemistry, 52(7), 1013(1980). [14] L.Larai, Y. Harek, A. Reguig, M.M. Mostafa, Journal of Serbian chemical society, 68(2), 85(2003). [15] W.P. Griffith, S.I. Mostafa, Polyhedron, 11(23), 2997(1992). [16] T. Priya Devi, R.K. Hemakumar Singh, Rasyan journal of chemistry, 3(2), 266(2010). [17] D. Rekha, N.Y. Sreedhar, P. Reddy Prasad, Global journal of science frontier research, 10(2), 21(2010). [18] A.A.Almeida. X. Jun, J.L.F.C. Lima., Talanta, 50(2), 253(1999). [19] A. Praveen Kumar, P. Raveendra Reddy, V. Krishna Reddy, Journal of Automated Methods Management Chemistry, 1(2007). [20] C.S. Kira, A.M. Sakuma, N. da Cruz Gouveia, Journal of Applied Pharmaceutical Science, 4 (5), 39(2014). [21] Z. Sun, P. Liang, Q. Ding, J. Cao, Hazard. Mater B, 137(2), 943(2006). [22] R.Galbeiro, S. Garcia, I. Gaubeur, Journal of trace elements in medicine and Biology, 28(2), 160(2014). [23] Q. Hu, G. Yang, Z. Huang, J.Yin. Analytical Science, 19(10), 1449(2003). [24] B. Saritha, T.S. Reddy, IOSR Journal of Applied Chemistry, 7(3), 22(2014). [25] H.Cyftci. A. Olcucu, T. Cyftci, International Journal of Science&Technology, 2(2), 105(2007). [26] Q. Hu, G. Yang, Z. Huang, J. Yin. Buliton of Korean Chemical Society, 25(4), 545(2004).
Copyright
Copyright © 2024 S. Hemanth Phani Kumar, Dasari Rekha. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET59356
Publish Date : 2024-03-24
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online