

Ijraset Journal For Research in Applied Science and Engineering Technology
Revolutionizing Drug Discovery: A Comprehensive Review of Computer-Aided Drug Design Approaches
Authors: Bharti Oli, Dr. Virender Kaur, Dr. Tirath Kumar
DOI Link: https://doi.org/10.22214/ijraset.2024.63563
Certificate: View Certificate
Abstract
Computer-Aided Drug Design (CADD) has significantly advanced the drug discovery process, offering tools to enhance efficiency and reduce costs. This review explores essential CADD methodologies, including molecular docking, virtual screening, ADMET profiling, homology modeling, and Quantitative Structure-Activity Relationship (QSAR) models. Molecular docking predicts interactions between drugs and targets, while virtual screening evaluates large compound libraries to identify promising candidates. ADMET profiling assesses pharmacokinetic and toxicological properties early in development. Homology modeling constructs three-dimensional protein models to aid target identification, and QSAR models predict biological activities based on chemical structures. These integrated approaches streamline drug development, providing a robust framework for modern pharmaceutical research.
Introduction
I. INTRODUCTION
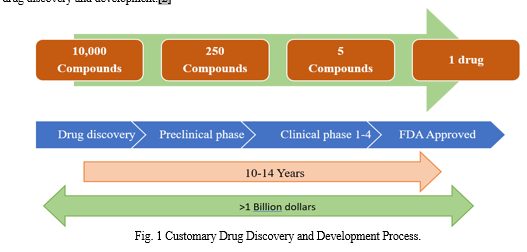
The process of bringing a novel medication to market is costly and time-consuming. The normal time frame for discovering and developing a medicine is ten to fifteen years, costing about US$ 800 million. In the 1990s, several advances were undertaken in utilizing high-throughput and combinatorial screening technologies that accelerated the process of finding new drugs. These technologies were widely adopted because they allowed for the rapid synthesis and screening of large libraries; nonetheless, little progress was made in the direction of the discovery of new molecular entities, and no significant breakthrough was achieved.
Modern computer techniques, chemical synthesis, and biological research were used to expedite the discovery process; this combinational approach broadened the field of discovery. The use of computers in drug discovery eventually came to be known as computer-aided drug design, or CADD.[1]
To cut down on time, expense, and risk-related factors, the computer-aided drug design (CADD) method is being increasingly used as a new drug design methodology. It has been shown that the application of CADD approaches can result in a 50% reduction in the cost of drug discovery and development.[2]

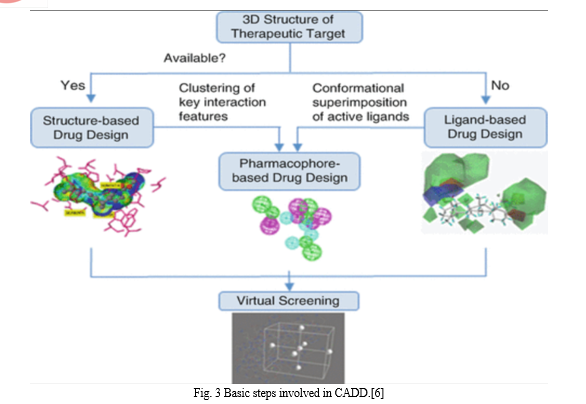
Two primary approaches to drug design with CADD are as follows:
- Structure-Based Drug Design
- Ligand Based Drug Design
- Structure-based Drug Design
The three-dimensional structure of the therapeutic target proteins and an examination of the binding site cavity serve as the foundation for structure-based drug design, or SBDD. Its rapid and accurate approach to finding and refining promising molecules has contributed to our understanding of disease at the molecular level. Among the methods commonly employed in SBDD are molecular docking, molecular dynamics (MD) simulations, and structure-based virtual screening (SBVS). These methods have several uses, such as assessing protein-ligand interactions, binding energetics, and receptor conformational changes caused by ligand binding.[7]
SBDD locates potentially innovative drug candidates by using knowledge about biological targets. Consequently, SBDD signifies a noteworthy advancement in computational techniques utilized in many fields such as biochemistry, pharmaceutical chemistry, statistics, and biophysics.[1] SBDD attempts to anticipate the ligands' binding affinity, or Gibbs free energy of binding (ΔGbind), by modeling the interactions between ligands and binding sites.[3]
Currently available on the market are a number of drugs that were discovered through SBDD. One of the most prominent instances of SBDD's efficacy is the utilization of FDA-approved drugs that suppress the HIV-1 virus. A number of failed attempts are also documented; for example, the antidepressant RPX00023 was supposed to operate as an agonist on receptor 5-HT1A, but it actually inhibited the receptor.[8]
The sequential procedure of SBDD entails gathering data piece by piece. In silico studies are performed starting with a known target structure in order to identify potential ligands. Following these molecular modeling procedures, the most promising compounds are subsequently produced. Evaluations of biological characteristics, such as potency, affinity, and efficacy, are then carried out via a number of experimental platforms.[9]
2. Ligand-based Drug Design
The "ligand-based drug design" (LBDD) approach to drug development uses a set of chemical structures of ligands that conform to a target without the target structure. In the LBDD process, pharmacophore models, quantitative structure-activity relationships (QSAR), and molecular similarity approaches are frequently used.[10]
According to Hendrickson (1991), the fundamental tenet of LBDD is that molecules with similar structures are likely to have similar properties. The first step in LBDD is the development and retrieval of small molecule libraries.[11]
When a 3D structure is not available for experimentation, ligand-based drug design techniques can be helpful. For ligand-based approaches, natural products or substrate analogues that bind with the target molecule and provide the desired pharmacological effect can also be employed instead of well-known ligand molecules. The QSAR method and pharmacophore modeling are the most widely used techniques for ligand-based drug design.
II. QSAR (QUANTITATIVE STRUCTURE-ACTIVITY RELATIONSHIP)
The association between a certain chemical or biological process and the chemical structures of a collection of chemicals is measured using a computer method known as QSAR. The basic premise of the QSAR approach states that similar structural or physiochemical characteristics lead to similar activity.[12]
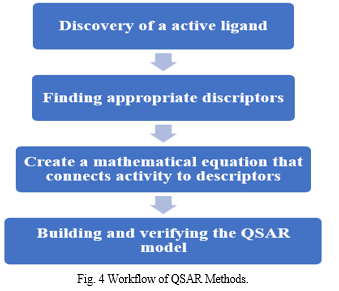
QSAR models are built for computational drug design, activity prediction, and toxicity prediction. QSAR is the quantitative relationship between properties discovered in chemical research and biological activity.
Biological activity = f (physicochemical parameter)[13]
The concept that changes in a collection of chemicals' molecular structures can be quantitatively linked to changes in their biological activity is the basis of the QSAR technique. Because of this, every molecular activity and function involved in biology is linked to a specific molecular descriptor, and the relative contributions of those descriptors to the biological effect may be ascertained using certain regression techniques.[14]
Six categories can be used to categorize QSAR based on the descriptors' methods of extraction:
- 1D-QSAR, which studies the connection between global molecular properties like pKa and logP and biological activities,
- 2D-QSAR, which correlates biological activities using structural patterns like connection indices and 2D-pharmacophores.
- 3D-QSAR, This technique studies the connection between ligands' noncovalent interaction fields and biological activity.
- 4D-QSAR, a 3D-QSAR extension containing an ensemble of ligand configurations.
- 5D-QSAR, an extension of 4D-QSAR that contains multiple induced-fit models; and
- 6D-QSAR, an addition to 5D-QSAR that includes more solvation models.[7]
A. Hologram Quantitative Structure-activity Relationship (HQSAR)
Hologram QSAR is a novel QSAR technique that does not require precise 3D information about the ligands. By using this method, the molecule divides into a molecular fingerprint that represents the frequency with which various kinds of molecular fragments occur. In other words, the minimum and maximum lengths of the pieces are determined by the size of the fragment to be included in the hologram fingerprint.
B. Comparative Molecular Field Analysis (CoMFA)
Comparative molecular field analysis, or CoMFA, is a helpful new technique for understanding the relationship between structure and activity. Work on CoMFA, a well-known 3D QSAR approach, began in the 1970s. It gives an explanation of the ligands' steric and electrostatic values as well as ClogP values, which show that a solvent-repellent restricts the ligands.
C. Comparative Molecular Similarity Indices Analysis (CoMSIA)
Comparative Molecular Similarity Indices Analysis (CoMSIA) is recognized as one of the most recent 3DQSAR approaches. It is commonly used to uncover common characteristics required for proper biological receptor binding throughout the drug discovery process. This method takes into account the steric and electrostatic characteristics, acceptors and donors of hydrogen bonds, and hydrophobic fields.[15]
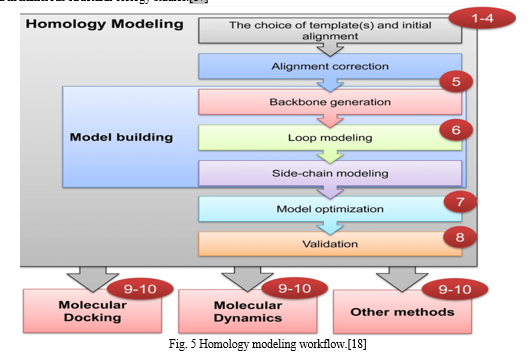
III. HOMOLOGY MODELLING
Homology modeling, sometimes referred to as comparative modeling, is one of the computational structure prediction methods used to determine a protein's three-dimensional structure from its amino acid sequence using a template. It is believed that homology modeling is the most accurate method for predicting computational structures.
Homology modeling is based on two important discoveries. A protein's 3D structure is mostly determined by its amino acid sequence. Second, the structure of proteins is more conserved and evolves far more slowly than sequence during evolution. Because of this, sequences that are similar fold into identical structures, and even sequences that are unrelated take on similar structures.[16]
In recent years, access to theoretical three-dimensional (3D) structures of molecular targets has been made possible through the growing popularity of homology modeling. Thus far, this technique has yielded several three-dimensional protein models that have been used in numerous structural biology studies.[17]
IV. MOLECULAR DOCKING
Placing molecules in the optimal places to interact with a receptor is known as Molecular Docking. When molecules come together to form a stable complex in a matter of minutes, a phenomenon known as docking takes place in the cell, as seen in Figure 7.[19]
Through the application of the molecular docking technique, we can model the atomic-level interaction between a small molecule and a protein, providing insight into fundamental biochemical processes and elucidating the behavior of small molecules within the binding sites of target proteins.[20]
The protein-ligand combination shows maximal interaction in the configuration identified by molecular docking with the least amount of energy. It also recognizes different protein targets and their inhibitors and then synthesizes the appropriate compounds or ligands to bind to them. This process is affected by a wide range of factors, including intramolecular forces (bond length, bond angle) and intermolecular forces (electrostatic, van der Waals, and others). Among the docking types are protein-protein, protein-ligand, lock-key, fitting, and flexible docking.[21]
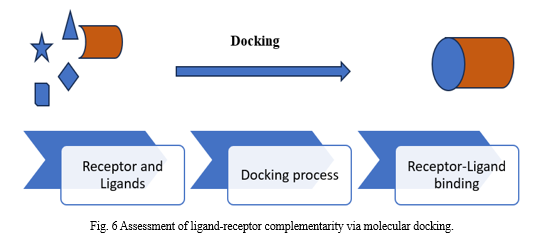
A. Docking Methodologies
- Rigid ligand and Rigid Receptor Docking: If the ligand and receptor are both thought of as rigid bodies, then there are just three translational and three rotational degrees of freedom to take into account in the search space. In this case, ligand flexibility might be addressed by using a pre-computed set of ligand conformations, or by allowing for a certain amount of atom-to-atom overlap between the protein and ligand.
- Flexible Ligand and Rigid Receptor Docking: For systems that follow the induced fit paradigm, it is important to consider the flexibilities of both the ligand and the receptor. In such circumstances, a minimum energy perfect-fit combination is produced by the ligand and receptor both changing their conformations. The cost is quite large when the receptor is also flexible. Therefore, the conventional approach is to consider the ligand as flexible during docking while maintaining the rigidity of the receptor, which also includes a trade-off between computing time and accuracy.
- Flexible Ligand and Flexible Receptor Docking: Teague examined the data demonstrating a direct connection between protein intrinsic mobility and ligand binding behavior. Taking into account the flexibility of the receptor is a significant issue in the field of docking. At now, there exist multiple approaches to implement the receptor flexibility. The simplest technique, called "soft-docking," consists of lowering the scoring function's van der Waals repulsive energy term to allow for a certain degree of ligand-receptor atom-to-atom overlap.[20]
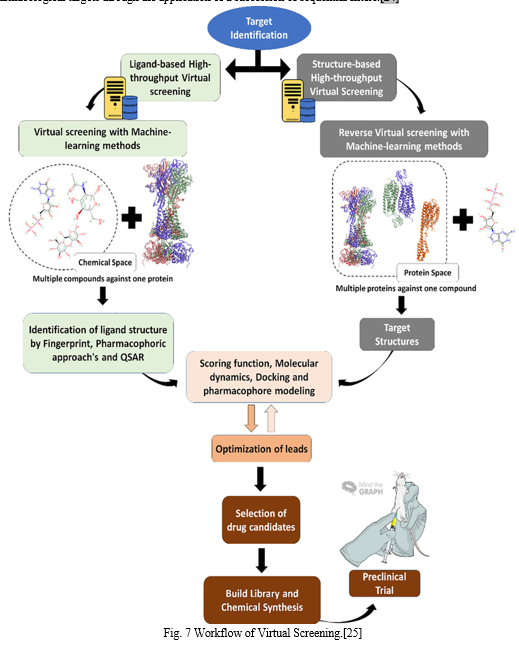
IV. VIRTUAL SCREENING
Due to the fact that screening billions of chemicals biologically still necessitates a large amount of experimental work, computer-aided drug design technologies have become popular as practical substitutes. In recent years, virtual screening has become a lucrative and dynamic technique in the pharmaceutical business for finding new medications, or "hits".[22]
These days, Virtual screening is an essential part of the drug discovery procedure. Usually, it takes the form of a hierarchical workflow that ranks potentially active chemicals either simultaneously or sequentially using a variety of filtering strategies.
Virtual screening techniques are often separated into two main groups:
- Strategies based on structure, emphasizing the complementary nature of the binding pocket between the ligand and the target;
- Strategies based on ligands, depending on how much new compounds resemble active compounds that are already on the market.[23]
Virtual screening is a thorough, knowledge-based approach to searching through compound databases in search of novel compounds and chemotypes with the biological activity required to take the place of already used ligands. Virtual screening is often described as a methodical procedure that selects and narrows down a set of lead-like hits with the potential to exhibit biological activity against certain pharmacological targets through the application of a succession of sequential filters.[24]
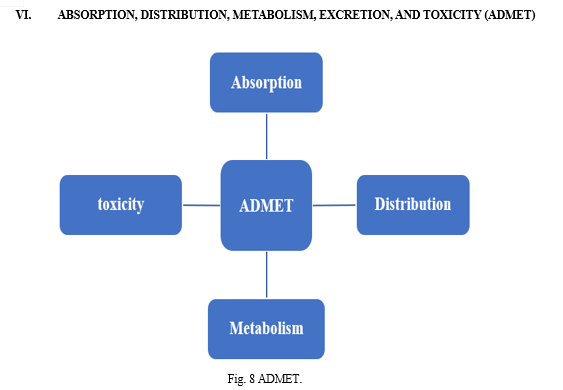
The search and development of novel medications is a labor-intensive and financially demanding process. This method includes lead optimization, disease selection, target discovery and validation, and preclinical and clinical trials. Two main elements contribute to drug failure: safety and ineffectiveness. Therefore, at every stage of the drug development process, chemical qualities relating to absorption, distribution, metabolism, excretion, and toxicity (ADMET) are crucial. Finding efficient molecules with enhanced ADMET properties is therefore crucial. Drug likeness provides a useful framework for the early stages of drug development.[26]
Numerous compounds can be evaluated prior to production and testing by using chemical structures to anticipate ADMET characteristics. It has recently been shown that ADMET characteristics can be predicted theoretically with reasonable accuracy. The primary barrier to modeling ADMET attributes has been the lack of adequate high-quality experimental data to build reliable models.[27]
For a potent chemical to be effective as a drug, it must reach its target in the body at a high enough concentration and stay there in a bioactive form for the expected biologic processes to occur. Absorption, distribution, metabolism, and excretion (ADME) are assessed at ever earlier stages of the discovery phase in the drug development process, when there are a large number of possible compounds but limited physical sample availability. Computer simulations are acceptable replacements for experiments in that context. Here we present the SwissADME web tool, a new resource that offers free access to a range of fast and accurate predictive models for drug-likeness, pharmacokinetics, physicochemical qualities, and medicinal chemistry friendliness.[28]
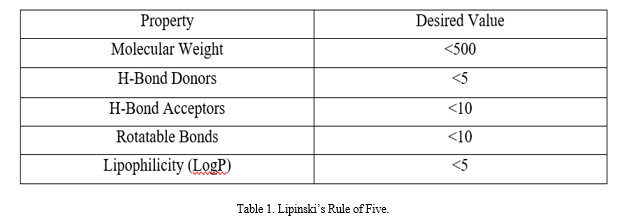
VII. LIPINSKI’S RULE OF FIVE
Lipinski's rule of five (Ro5) has been the standard "rule of thumb" for assessing traits that are similar to drugs for almost 20 years. It is a commonly accepted method to predict the ADME ("absorption, distribution, metabolism, and excretion") performance of pharmaceuticals, especially oral treatments.
Lipinski looked back at 2,245 compounds at the beginning of Phase II development programs in order to establish the Ro5. Lipinski identified the usual physicochemical properties of the selected compounds.[29]
This criterion has been justified by the idea that creating compounds, especially compound libraries, that are too large, floppy, or apolar may reduce their likelihood of exhibiting oral bioavailability and other desired pharmacological properties.
The Ro5 is a widely accepted guideline for the development of orally bioavailable small-molecule drugs, but there are many compounds that break it that make good drug candidates as well, and it is increasingly accepted that efforts to develop new drugs should not be restricted to the Ro5 chemical space.[30]
Conclusion
Computer-Aided Drug Design (CADD) represents a pivotal advancement in the field of pharmaceutical research, offering powerful tools to streamline and enhance the drug discovery process. This review has highlighted the key methodologies within CADD, including molecular docking, virtual screening, ADMET profiling, homology modeling, and QSAR models. Each of these approaches contributes uniquely to identifying, evaluating, and optimizing potential drug candidates. By integrating these computational techniques, researchers can predict drug-target interactions, assess pharmacokinetic properties, and construct accurate models of target proteins, thereby accelerating the development of effective and safe therapeutic agents. As CADD continues to evolve, its role in revolutionizing drug discovery becomes increasingly apparent, promising a future where new drugs can be developed more efficiently and precisely. This integration of technology into drug design not only optimizes the research process but also holds the potential to bring innovative treatments to patients more rapidly.
References
[1] M. H. Baig, K. Ahmad, G. Rabbani, M. Danishuddin, and I. Choi, “Computer Aided Drug Design and its Application to the Development of Potential Drugs for Neurodegenerative Disorders,” Curr. Neuropharmacol., vol. 16, no. 6, pp. 740–748, Jul. 2018, doi: 10.2174/1570159X15666171016163510. [2] S. Surabhi and B. K. Singh, “COMPUTER AIDED DRUG DESIGN: AN OVERVIEW,” J. Drug Deliv. Ther., vol. 8, pp. 504–509, Sep. 2018, doi: 10.22270/jddt.v8i5.1894. [3] Y. Chang, B. A. Hawkins, J. J. Du, P. W. Groundwater, D. E. Hibbs, and F. Lai, “A Guide to In Silico Drug Design,” Pharmaceutics, vol. 15, no. 1, p. 49, Dec. 2022, doi: 10.3390/pharmaceutics15010049. [4] B. Shaker, S. Ahmad, J. Lee, C. Jung, and D. Na, “In silico methods and tools for drug discovery,” Comput. Biol. Med., vol. 137, p. 104851, Oct. 2021, doi: 10.1016/j.compbiomed.2021.104851. [5] M. Roney and M. F. F. Mohd Aluwi, “The importance of in-silico studies in drug discovery,” Intell. Pharm., Feb. 2024, doi: 10.1016/j.ipha.2024.01.010. [6] D. B. Singh, M. K. Gupta, R. K. Kesharwani, M. Sagar, S. Dwivedi, and K. Misra, “Molecular drug targets and therapies for Alzheimer’s disease,” Transl. Neurosci., vol. 5, no. 3, pp. 203–217, Sep. 2014, doi: 10.2478/s13380-014-0222-x. [7] A. B. Gurung, M. A. Ali, J. Lee, M. A. Farah, and K. M. Al-Anazi, “An Updated Review of Computer-Aided Drug Design and Its Application to COVID-19,” BioMed Res. Int., vol. 2021, p. 8853056, Jun. 2021, doi: 10.1155/2021/8853056. [8] M. Batool, B. Ahmad, and S. Choi, “A Structure-Based Drug Discovery Paradigm,” Int. J. Mol. Sci., vol. 20, no. 11, p. 2783, Jun. 2019, doi: 10.3390/ijms20112783. [9] L. G. Ferreira, R. N. dos Santos, G. Oliva, and A. D. Andricopulo, “Molecular Docking and Structure-Based Drug Design Strategies,” Molecules, vol. 20, no. 7, pp. 13384–13421, Jul. 2015, doi: 10.3390/molecules200713384. [10] J. Ye, X. Yang, and C. Ma, “Ligand-Based Drug Design of Novel Antimicrobials against Staphylococcus aureus by Targeting Bacterial Transcription,” Int. J. Mol. Sci., vol. 24, no. 1, Art. no. 1, Jan. 2023, doi: 10.3390/ijms24010339. [11] V. Sharma, S. Wakode, and H. Kumar, “Structure- and ligand-based drug design: concepts, approaches, and challenges,” 2021, pp. 27–53. doi: 10.1016/B978-0-12-821748-1.00004-X. [12] C. Acharya, A. Coop, J. E. Polli, and A. D. MacKerell, “Recent Advances in Ligand-Based Drug Design: Relevance and Utility of the Conformationally Sampled Pharmacophore Approach,” Curr. Comput. Aided Drug Des., vol. 7, no. 1, pp. 10–22, Mar. 2011. [13] V. Bastikar, A. Bastikar, and P. Gupta, “Quantitative structure–activity relationship-based computational approaches,” Comput. Approaches Nov. Ther. Diagn. Des. Mitigate SARS-CoV-2 Infect., pp. 191–205, 2022, doi: 10.1016/B978-0-323-91172-6.00001-7. [14] L. Abdel-Ilah, E. Veljovic, L. Gurbeta, and A. Badnjevic, “Applications of QSAR Study in Drug Design,” Int. J. Eng. Res. Technol., vol. 6, no. 6, Jun. 2017, doi: 10.17577/IJERTV6IS060241. [15] A. Wadood, N. Ahmed, L. Shah, A. Ahmad, H. Hassan, and S. Shams, “In-silico drug design: An approach which revolutionarised the drug discovery process,” OA Drug Des. Deliv., vol. 1, Sep. 2013, doi: 10.13172/2054-4057-1-1-1119. [16] M. T. Muhammed and E. Aki?Yalcin, “Homology modeling in drug discovery: Overview, current applications, and future perspectives,” Chem. Biol. Drug Des., vol. 93, no. 1, pp. 12–20, Jan. 2019, doi: 10.1111/cbdd.13388. [17] T. C. C. França, “Homology modeling: an important tool for the drug discovery,” J. Biomol. Struct. Dyn., vol. 33, no. 8, pp. 1780–1793, Aug. 2015, doi: 10.1080/07391102.2014.971429. [18] “Fig 1. Homology-modeling workflow. Numbers show the steps where tips...,” ResearchGate. Accessed: Jul. 03, 2024. [Online]. Available: https://www.researchgate.net/figure/Homology-modeling-workflow-Numbers-show-the-steps-where-tips-are-applicable_fig1_340396142 [19] K. Raval and T. Ganatra, “Basics, types and applications of molecular docking: A review,” IP Int. J. Compr. Adv. Pharmacol., vol. 7, pp. 12–16, Mar. 2022, doi: 10.18231/j.ijcaap.2022.003. [20] X.-Y. Meng, H.-X. Zhang, M. Mezei, and M. Cui, “Molecular Docking: A powerful approach for structure-based drug discovery,” Curr. Comput. Aided Drug Des., vol. 7, no. 2, pp. 146–157, Jun. 2011. [21] V. Kandi, A. Vundecode, T. Godalwar, S. Dasari, S. Vadakedath, and V. Godishala, “The Current Perspectives in Clinical Research: Computer-Assisted Drug Designing, Ethics, and Good Clinical Practice,” Borneo J. Pharm., vol. 5, pp. 161–178, May 2022, doi: 10.33084/bjop.v5i2.3013. [22] A. Reddy, S. Pati, P. K. Potukuchi, P. H N, and G. N. Sastry, “Virtual Screening in Drug Discovery - A Computational Perspective,” Curr. Protein Pept. Sci., vol. 8, pp. 329–51, Sep. 2007, doi: 10.2174/138920307781369427. [23] T. B. Kimber, Y. Chen, and A. Volkamer, “Deep Learning in Virtual Screening: Recent Applications and Developments,” Int. J. Mol. Sci., vol. 22, no. 9, p. 4435, Apr. 2021, doi: 10.3390/ijms22094435. [24] A. Lavecchia and C. Giovanni, “Virtual Screening Strategies in Drug Discovery: A Critical Review,” Curr. Med. Chem., vol. 20, no. 23, pp. 2839–2860, Jun. 2013, doi: 10.2174/09298673113209990001. [25] A. Alberto, N. Ferreira, R. Soares, and L. Alves, “Molecular Modeling Applied to the Discovery of New Lead Compounds for P2 Receptors Based on Natural Sources,” Front. Pharmacol., vol. 11, Sep. 2020, doi: 10.3389/fphar.2020.01221. [26] L. Guan et al., “ADMET-score – a comprehensive scoring function for evaluation of chemical drug-likeness †Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00472b,” MedChemComm, vol. 10, no. 1, pp. 148–157, Nov. 2018, doi: 10.1039/c8md00472b. [27] D. Cao, J. Wang, R. Zhou, Y. Li, H. Yu, and T. Hou, “ADMET Evaluation in Drug Discovery. 11. PharmacoKinetics Knowledge Base (PKKB): A Comprehensive Database of Pharmacokinetic and Toxic Properties for Drugs,” J. Chem. Inf. Model., vol. 52, no. 5, pp. 1132–1137, May 2012, doi: 10.1021/ci300112j. [28] A. Daina, O. Michielin, and V. Zoete, “SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules,” Sci. Rep., vol. 7, p. 42717, Mar. 2017, doi: 10.1038/srep42717. [29] T. K. Karami, S. Hailu, S. Feng, R. Graham, and H. J. Gukasyan, “Eyes on Lipinski’s Rule of Five: A New ‘Rule of Thumb’ for Physicochemical Design Space of Ophthalmic Drugs,” J. Ocul. Pharmacol. Ther., vol. 38, no. 1, pp. 43–55, Jan. 2022, doi: 10.1089/jop.2021.0069. [30] M. Egbert, A. Whitty, G. M. Keser?, and S. Vajda, “Why some targets benefit from beyond rule of five drugs,” J. Med. Chem., vol. 62, no. 22, pp. 10005–10025, Nov. 2019, doi: 10.1021/acs.jmedchem.8b01732.
Copyright
Copyright © 2024 Bharti Oli, Dr. Virender Kaur, Dr. Tirath Kumar . This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET63563
Publish Date : 2024-07-05
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online