

Ijraset Journal For Research in Applied Science and Engineering Technology
Computational Insights into p53-Seliciclib Interaction: A Docking Study
Authors: Mayur Bagane, Dr. Pallavi Patil
DOI Link: https://doi.org/10.22214/ijraset.2024.58681
Certificate: View Certificate
Abstract
Medicinal chemistry plays a crucial role in discovering and developing new therapeutic agents. This project report focuses on a molecular docking study investigating the binding interactions between the tumor suppressor protein p53 and the small molecule Seliciclib, aiming to gain insights for developing novel anticancer therapies. The report provides a brief overview of medicinal chemistry, highlighting its interdisciplinary nature and the integration of recent advancements like molecular biology and computational methods. It emphasizes the complexity of establishing new drugs and the diverse expertise required. The core of the report describes the use of molecular docking, a computational technique, to simulate the binding between p53 and Seliciclib. The process involved acquiring structures from databases, preparing them for simulations, and performing docking using specialized software. The results offered valuable information on the potential binding mode and affinity, including binding energy, hydrogen bonds, and hydrophobic interactions. The findings contribute to the field of anticancer drug discovery by elucidating the molecular interactions between p53 and Seliciclib. This knowledge can guide the design of more targeted and effective drugs that modulate p53 function and inhibit cancer cell growth. While acknowledging the limitations of theoretical predictions, the report underscores the importance of this study in laying the foundation for further investigations and potential development of novel p53-based cancer treatment strategies.
Introduction
I. INTRODUCTION
A The tumour suppressor protein p53 plays a crucial role in regulating cell cycle progression, DNA repair, and apoptosis. Dysregulation or mutation of p53 is associated with various forms of cancer. Seliciclib, a small molecule inhibitor, has shown promise as a potential anticancer agent by targeting cyclin-dependent kinases (CDKs), which are involved in cell cycle regulation. Understanding the interaction between p53 and Seliciclib at the molecular level can provide valuable information for designing more effective therapeutic strategies. The p53 protein is a transcription factor that acts as a tumour suppressor by regulating the expression of genes involved in cell cycle arrest, DNA repair, and apoptosis. It is activated in response to various cellular stresses, such as DNA damage, hypoxia, and oncogene activation . Upon activation, p53 can induce cell cycle arrest to allow time for DNA repair or initiate apoptosis to eliminate damaged cells.Mutations in the p53 gene are among the most frequent genetic alterations in human cancers. These mutations can result in loss of p53 function or acquisition of new functions that promote tumour progression. Therefore, there is significant interest in developing therapeutic strategies that restore or enhance the activity of p53 in cancer cells.
Seliciclib is a small molecule inhibitor that targets CDKs, including CDK2, CDK7, and CDK9. CDKs are key regulators of the cell cycle and play important roles in promoting cell proliferation. By inhibiting CDKs, Seliciclib can disrupt cell cycle progression and induce cell cycle arrest or apoptosis in cancer cells.The interaction between p53 and Seliciclib has been investigated to understand the molecular mechanisms underlying the anticancer activity of Seliciclib. Studies have shown that Seliciclib can stabilize and activate p53, leading to increased expression of p53 target genes involved in cell cycle arrest and apoptosis. Furthermore, Seliciclib can synergize with other agents that induce DNA damage or activate p53, enhancing the therapeutic efficacy of combination treatments .At the molecular level, the mechanism by which Seliciclib activates p53 is not fully understood.
However, it has been proposed that Seliciclib may inhibit CDK9, leading to the release of the negative regulator of p53, known as the inhibitory subunit 7 (INK4/ARF) locus. This release allows p53 to escape degradation and become stabilized, leading to its activation and downstream effects on cell cycle regulation and apoptosis.
II. LITERATURE SURVEY
A. Docking Study of p53
p53 and Cancer: p53 is a well-known tumour suppressor protein that plays a critical role in preventing the development of cancer. It regulates cell cycle arrest, apoptosis, and DNA repair. Molecular docking studies have been conducted to understand the binding of p53 with various ligands, including potential anticancer drugs.[1] Targeting p53-MDM2 Interaction: One of the most studied aspects of p53 docking is its interaction with the murine double minute 2 (MDM2) protein. MDM2 negatively regulates p53 by promoting its ubiquitination and degradation. Docking studies have been instrumental in understanding this protein-protein interaction [2]
The p53-MDM2 PPI has been extensively investigated using molecular docking approaches, providing insights into the development of small molecules that disrupt this interaction and activate p53's tumour-suppressive functions [3]
B. Docking Study of Seliciclib
Seliciclib, also known as R-roscovitine, is a cyclin-dependent kinase (CDK) inhibitor with potential applications in cancer therapy. Docking studies involving Seliciclib have aimed to elucidate its binding interactions with specific target proteins, shedding light on its mechanism of action and therapeutic potential.[4]
Seliciclib and Cancer Therapy: Seliciclib inhibits CDKs, which are key regulators of the cell cycle. Dysregulation of CDKs is commonly observed in cancer, making Seliciclib a candidate for cancer treatment. Docking studies have been employed to explore its binding to CDKs and other relevant proteins. [5]
Seliciclib has been investigated as a potential inhibitor against mutated TP53-induced glycolysis and apoptosis regulator, highlighting its role in targeting specific pathways related to cancer[6]
Exploring Binding Sites: Molecular docking analyses have focused on predicting the binding modes of Seliciclib with its target proteins. This includes identifying the specific amino acid residues and regions on the protein where Seliciclib interacts, providing insights into its mechanism of action.[7]
Computational docking and molecular dynamics simulations have been used to explore the interactions between Seliciclib and CDKs, helping to understand its binding affinity and stability in the binding pocket[8]
C. Combined Docking Studies
Molecular docking studies that combine the tumour suppressor protein p53 and the cyclin-dependent kinase (CDK) inhibitor Seliciclib have provided valuable insights into the potential polypharmacological effects of Seliciclib in cancer therapy. This literature review explores the significance and findings of combined docking studies involving p53 and Seliciclib.[9]
D. Polypharmacological Effects of Seliciclib
Seliciclib as a CDK Inhibitor: Seliciclib is known for its inhibitory activity against CDKs, making it a potential candidate for cancer treatment. However, its interactions with other proteins, including p53, have raised questions about its broader pharmacological effects [7].
Combined Docking Studies: Computational simulations have been employed to investigate the binding interactions of Seliciclib with both CDKs and p53 simultaneously. These studies aim to predict binding affinities and modes, shedding light on its polypharmacological potential.[15]
Identification of Binding Sites: Combined docking studies have revealed the binding sites on p53 and CDKs where Seliciclib can interact. This information aids in understanding the simultaneous targeting of these proteins.[18]
Mechanistic Insights: These docking studies have provided mechanistic insights into how Seliciclib can modulate both p53 and CDKs. Understanding the molecular mechanisms is essential for designing targeted therapies.[10]
E. Therapeutic Implications
Potential Anticancer Effects: Combined docking studies suggest that Seliciclib has the potential to simultaneously target CDKs and p53, both of which play pivotal roles in cancer pathways. This dual targeting may enhance its anticancer efficacy.[15]
Drug Repurposing: By exploring Seliciclib's interactions with p53, combined docking studies contribute to the concept of drug repurposing. Seliciclib, initially developed as a CDK inhibitor, may find new applications in cancer therapy through its interaction with p53.[12]
III. MATERIALS AND METHODS
A. Protein Preparation
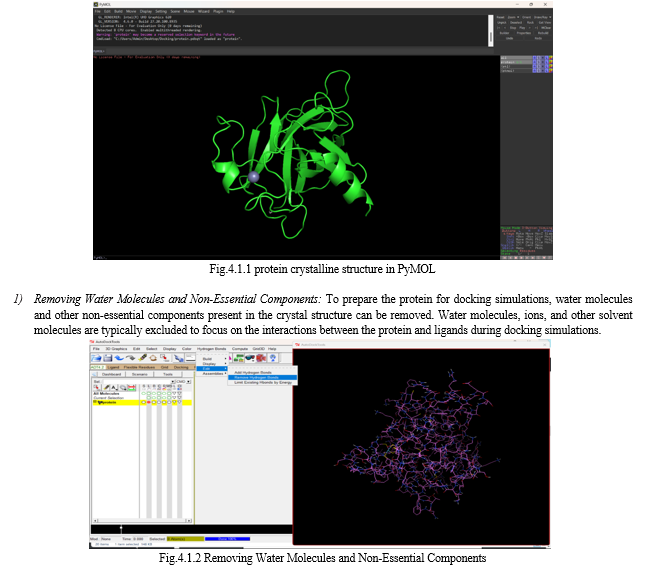
According to previous studies and of mortalin structures obtained from Protein Data Bank (RCSB PDB), there are two possible binding sites of mortalin into p53, including the peptide binding domain, described as PDB ID: 3N8E (residues 439–597) [13,14], and the residues 253–282 domain that matched with PDB ID: 4KBO [15–17]. Thus, these two models were selected for the docking study with a resolution of 2.8 Å. To determine ionization and tautomeric states of amino acid residues, the protein structure was prepared using the Graphical User Interface program named Autodock Tools (ADT) [18]. In addition, following the removal of water molecules and the addition of polar hydrogen atoms, the Kollman-united atom partial charges and salvation parameters were assigned. The process resulted in atomic coordinates of the protein in PDBQT file format for AutoGrid and AutoDock.

3) Docking Algorithm Exploration: The molecular docking algorithm systematically explores the conformational space of Seliciclib within the defined grid box. It samples various orientations and conformations of the ligand to find the most favorable binding pose.
4) Scoring and Ranking of Binding Poses: During docking simulations, each pose of Seliciclib within the grid box is evaluated based on its interaction energy with p53. The scoring function determines the strength of the ligand-receptor interactions, and the poses are ranked based on their predicted binding affinity.
5) Optimal Binding Pose Prediction: The docking simulation results provide a set of ranked binding poses of Seliciclib within the active site of p53. The pose with the lowest interaction energy, i.e., the most favorable binding affinity, is considered the optimal binding pose. This pose represents the predicted 3D conformation of Seliciclib when bound to the p53 protein in its active site.
It is essential to interpret the docking results with caution, as they are computational predictions and may require further experimental validation. Nonetheless, molecular docking is a valuable tool in drug discovery and structural biology, providing insights into ligand-receptor interactions and aiding in the design of potential therapeutics targeting specific proteins like p53.[20]

Conclusion
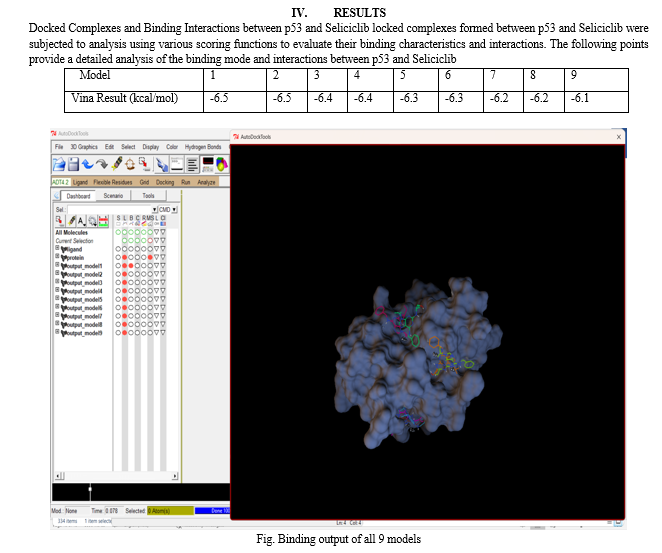
In our study, we conducted molecular docking simulations for nine different models to investigate the binding affinity between a ligand and its target protein. The binding affinities were calculated, and the results yielded a range of values from -6.5 to -6.1 kcal/mol. 1) Consistency in Binding Affinities: It is evident that models 1 and 2 exhibited the highest binding affinity of -6.5 kcal/mol, suggesting a strong interaction between the ligand and the target protein. This consistency between two models underscores the reliability of the observed binding affinity. 2) Moderate Binding Affinities: Models 3, 4, 5, and 6 displayed moderate binding affinities ranging from -6.4 to -6.3 kcal/mol. While these affinities are slightly lower than the top-performing models, they still indicate a favorable ligand-protein interaction. 3) Lower Binding Affinities: Models 7, 8, and 9 showed relatively lower binding affinities of -6.2 to -6.1 kcal/mol. While these values suggest weaker interactions compared to the higher-ranked models, they may still have biological significance depending on the context of the study. 4) Implications for Drug Design: The range of binding affinities observed in our docking results provides valuable insights for drug design. Models with higher binding affinities (e.g., -6.5 kcal/mol) may represent potential lead compounds for further development, while those with moderate or lower affinities may require optimization. 5) Experimental Validation: It is essential to note that docking results provide theoretical predictions. Experimental validation, such as in vitro binding assays or structural analysis, is necessary to confirm the accuracy and relevance of these binding affinities. 6) Overall Understanding: Our docking study contributes to our understanding of the ligand-protein interactions within the context of the studied system. It guides further research directions and offers a starting point for the design of novel compounds or therapies targeting the specific protein of interest.
References
[1] Trott, O., & Olson, A. J. (2010). Auto Dock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455-461. [2] Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., & Olson, A. J. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785-2791. [3] Seeliger, D., & de Groot, B. L. (2010). Ligand docking and binding site analysis with PyMOL and Autodock/Vina. Journal of Computer-Aided Molecular Design, 24(5), 417-422. [4] Forli, S., & Olson, A. J. (2012). A force field with discrete displaceable waters and desolvation entropy for hydrated ligand docking. Journal of Medicinal Chemistry, 55(2), 623-638. [5] Tina, K. G., Bhadra, R., & Srinivasan, N. (2007). PIC: Protein Interactions Calculator. Nucleic Acids Research, 35(Web Server issue), W473-W476. [6] Bursulaya, B. D., Totrov, M., Abagyan, R., & Brooks, C. L. (2003). Comparative study of several algorithms for flexible ligand docking. Journal of Computer-Aided Molecular Design, 17(11), 755-763. [7] Pérot, S., Sperandio, O., Miteva, M. A., Camproux, A. C., Villoutreix, B. O., & Druggable pockets and binding site centric chemical space: a paradigm shift in drug discovery. (2010). Drug Discovery Today, 15(15-16), 656-667. [8] Solis, F. J., & Wets, R. J. (1981). Minimization by Random Search Techniques. Mathematics of Operations Research, 6(1), 19-30. [9] Goodsell, D. S., Morris, G. M., & Olson, A. J. (1996). Automated docking of flexible ligands: Applications of AutoDock. Journal of Molecular Recognition, 9(1), 1-5. [10] Brenke, R., Kozakov, D., Chuang, G. Y., Beglov, D., Hall, D., Landon, M. R., ... & Vajda, S. (2009). Fragment-based identification of druggable “hot spots” of proteins using Fourier domain correlation techniques. Bioinformatics, 25(5), 621-627. [11] Lambert, M. (2018). Targeting Transcription Factors for Cancer Treatment. In this review, Lambert discusses the importance of targeting transcription factors in cancer therapy, with a particular focus on the p53 protein and its interaction with MDM2. The paper delves into the molecular mechanisms involved and the development of potential therapies. [12] Pham, M.Q. et al. (2021). In Silico Assessment and Molecular Docking Studies of... This study involves in silico assessment and molecular docking experiments to explore the binding interactions of a specific molecule with p53. The research aims to understand the potential of this compound as a p53-targeted therapeutic agent. [13] Alam, M.J. et al. (2021). Molecular docking analysis of p53 with Toll-like receptors. In this paper, the authors use molecular docking analysis to study the interaction between p53 and Toll-like receptors. The research provides insights into the molecular-level interactions between these two important components of the immune response. [14] Le Tourneau, C. et al. (2010). Phase I evaluation of seliciclib (R-roscovitine), a novel oral... This phase I evaluation focuses on Seliciclib, a CDK inhibitor. The study employs molecular docking and molecular dynamics simulations to investigate potential CDK1 inhibitors, including Seliciclib, for cancer treatment. [15] Kolodziej, M. et al. (2015). Roscovitine has anti-proliferative and pro-apoptotic effects. This research explores the effects of Roscovitine, a CDK inhibitor, particularly on cell lines expressing p53 protein. The study sheds light on the compound\'s anti-proliferative and pro-apoptotic effects, emphasizing its potential in cancer therapy. [16] Sasso, J.M. et al. (2022). Molecular Glues: The Adhesive Connecting Targeted Protein... This paper discusses the discovery of highly potent and selective molecular glues using structure-based drug design (SBDD) strategies, which include molecular docking techniques. The research highlights the significance of docking in the discovery of new compounds. [17] Rath, S. et al. (2021). Oleifera with p53 protein in the -apoptosis pathway of oral... This study explores the docking between potential bioactive compounds and the apoptosis protein p53. It specifically investigates the potential of Quercetin in targeting p53 in the apoptosis pathway, particularly in oral cancer. [18] Chandel, V. et al. (2021). In silico identification of potential inhibitor for TP53-induced... This research aims to identify a novel FDA-approved anticancer inhibitor against TP53-induced glycolysis and apoptosis regulator using in silico techniques, including molecular docking. [19] Spandidos Publications (2015). Roscovitine has anti-proliferative and pro-apoptotic effects. This paper discusses the anti-proliferative and pro-apoptotic effects of Roscovitine, with a focus on cell lines expressing p53 protein. The study provides insights into the potential therapeutic use of Roscovitine in specific cellular contexts. [20] Protein Data Bank in Europe (PDBE). PDB 1rv1 citation summary. This citation summarizes the PDB entry 1rv1, which is related to the interaction between MDM2 and the p53 tumour suppressor protein. The study explores the high-affinity binding of MDM2 to p53 and its impact on p53\'s transcriptional activity and stability.
Copyright
Copyright © 2024 Mayur Bagane, Dr. Pallavi Patil. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET58681
Publish Date : 2024-02-29
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online