

Ijraset Journal For Research in Applied Science and Engineering Technology
Diabetes Insipidus: Diagnosis, Pathogenesis, and Management
Authors: Aashi Singh Bhadouria, Ishan Singh Bhadouria
DOI Link: https://doi.org/10.22214/ijraset.2023.54580
Certificate: View Certificate
Abstract
The inability to control one\'s own urine output due to diabetes insipidus (DI) is a serious medical condition that may affect a person at any age. Constant urine and thirst, particularly during sleep, might disrupt your sleep cycle. The researchers behind this study searched the PubMed database for any and all relevant publications on DI. Nephrogenic DI, which results from the kidney\'s inability to respond to arginine vasopressin (AVP), is often inherited, but central DI, which results from the brain\'s inability to produce enough AVP, might result from trauma, surgery, or tumours. Historically, treatments have included the administration of oxytocin and vasopressin-containing extracts from the posterior pituitary. In comparison to vasopressin, the synthetic vasopressin counterpart desmopressin has some benefits. Desmopressin in intranasal form was the first to market, but since then, oral tablet and melt versions have gained favour due to their ease. Amiloride, thiazide diuretics, indapamide, clofibrate, indomethacin, carbamazepine, and chlorpropamide are some more medicinal compounds. However, desmopressin remains the medicine of choice for treating DI. The review discusses desmopressin-targeted medicines, DI precipitants, and the physiology of water balance.
Introduction
I. INTRODUCTION
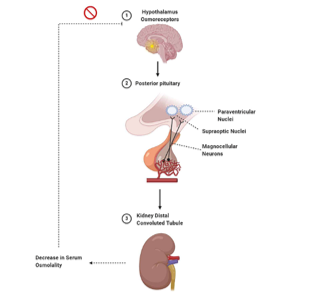
One in every 25,000 people has diabetes insipidus (DI), or 0.04% of the global population. Due to their low prevalence, the various DIs may be neglected in medical curricula and research efforts aimed at enhancing clinical treatment. Despite the fact that DI is an extremely uncommon endocrine disorder, the condition might negatively impact the patient's quality of life if left untreated. DI may affect people at any age and can affect either sex, while hereditary forms tend to show up sooner in life. DI may occur in four different ways: centrally, renally, fetally, and dipsogenically. If an adult has an osmolality of less than 300 mOsmol/kg in their urine and they pee more than 3-3.5 litres in a 24-hour period, they are considered to have DI. Dialysis patients often produce much more than 3.35–3.5 litres of urine in a 24-hour period. An overproduction of IGF-1 from the anterior pituitary gland is the root cause of diabetes insipidus. Antidiuretic hormone (ADH) increases urinary osmolyte content by activating the kidney. Osmoregulation and baroreceptor-mediated negative feedback play a significant role in controlling ADH secretion. Changes in plasma osmolality of less than 1 percent may be detected by the hypothalamic osmoreceptors. When blood osmolality increases, the posterior pituitary gland secretes antidiuretic hormone (ADH).
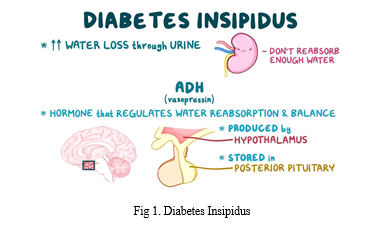
The baroreceptors, which become active in response to a decrease in blood volume, may be used to investigate a similar response. Due to the intrinsic volatility of blood volume, a volumetric fluctuation of 5-10% is necessary. Neurohypophysin II is involved in the bidirectional transfer of adrenocorticotropic hormone (ADH) between the hypothalamus to the posterior pituitary (where it is stored) (NPII). The water-soluble peptide hormone ADH is secreted into the circulation in response to changes in plasma osmolality or baroreceptor activation. Particularly, it interacts with aquaporin-2 receptors (AQP2) on the collecting duct's basolateral membrane. Binding to the receptor initiates the Gs-adenylyl cyclase pathway, which in turn increases intracellular cyclic AMP levels. Protein kinase A is activated by an increase in cAMP levels, and this leads to the phosphorylation of newly synthesised AQP2 channels. After AQP2 has been phosphorylated, it is transported to the cell's apical membrane. Results from experiments confirmed that, without AQP2, the renal collecting duct remained highly water-impermeable. Because of its role in reducing the quantity of water in kidney filtrate, AQP2 makes urine more concentrated. Damage to the collecting duct impedes water transport along an osmotic gradient from the nephron lumen into the collecting duct cells, resulting in dilute urine. Even while ADH may raise urine osmolality to over 1,200 mOsmol/kg, it can also decrease the rate of urine production to roughly 0.5 ml per min, or around 700-800 ml per day. Water homeostasis is accompanied by a decrease in the levels of adrenergic diuretic hormone (ADH) and apical plasma membrane aquaporin-2 (AQP2) channel proteins.
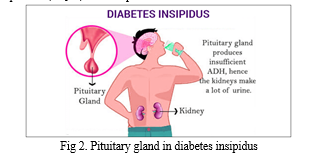
Diabetic insipidus is characterised by polyuria (>3 L/day) and polydipsia (>50 mL/kg body weight/24 hours), two forms of the water-loss syndrome known together as polyuria-polydipsia. In order to make a diagnosis of DI, osmotic diuresis disorders must first be ruled out (resulting from primary polydipsia). Another kind of DI may occur during pregnancy, although it is far less frequent, and it is called gestational DI. Hyponatremia of arginine vasopressin (AVP) is the root cause of central DI, and it may manifest in a person at birth or later in life. Nephrogenic DI develops when the kidneys stop responding to arginine vasopressin (AVP) as a result of pharmacological effects, electrolyte abnormalities, or mutations in the arginine vasopressin receptor 2 (AVPR2) or aquaporin 2 genes (AQP2). In primary polydipsia, there is a normal antidiuretic renal response and AVP secretion but nevertheless an abnormal amount of fluid intake. The end outcome is increased urination frequency (polyuria).
The failure to concentrate urine to its maximum capacity leads to water diuresis and is a common feature of all kinds of polyuria-polydipsia syndrome, regardless of origin. There are several kinds of therapy techniques, and it's vital to know the difference between them since some of them might be harmful if used incorrectly. Primary polydipsia, as well as partial types of central and nephrogenic DI, may be difficult to diagnose. Researched in this article are the many DI subtypes, what causes them, and the usual clinical presentation and clinical repercussions of central DI, such as polyuria. In addition to the normal radiological and laboratory procedures, we will also examine which specialized investigations (stimulation tests) are currently advised. Finally, we will discuss some of the in-hospital and out-of-hospital treatment options for central DI.
II. LITERATURE REVIEW
Ghousia Wadjida et.al., [16] Antibiotics other than penicillin, cephalosporin, sulfonamide, quinolone, and macrolide may also cause acute renal failure. Ten percent to fifteen percent of kidney biopsies in people with kidney failure of unknown cause show signs of acute renal failure. Patients' blood creatinine levels increase between 2 days to 44 days after pharmaceutical delivery, and much faster with repeated exposure to the same medicine.
IV administration of medicines such as sucrose, mannitol, and complex carbohydrates like dextran and hydroxyethyl starch have been related to AKI. On day 10 post-transplant, the gelatin group's serum creatinine levels was significantly lower than the HES-gelatin group's. Drug-induced AKI may typically be reversed when treatment is initiated, including correction of electrolyte imbalances and discontinuation of the offending medication.
Cynthia A Naughton, [12] The use of nephrotoxic medicines promotes the development of acute renal injury. Now more than ever, patients face the danger of renal toxicity due to the prevalence of several medical problems and the use of multiple treatment procedures, diagnostic tests, and therapeutic medications. Multiple pharmacological therapies increase the risk of drug-induced nephrotoxicities in patients. The need of taking precautions against these unwanted effects caused by several drugs such as aminoglycosides, cisplatin, cyclosporine, etc.
Devasmita Choudhury et.al.,[17] To date, there are no accurate serologic indications for detecting even mild renal injuries; nonetheless, kidney damage is often recognised before filtration is impeded, as shown by an increased blood urea nitrogen (BUN) and creatinine (Cr) level. It might be difficult to accurately assess the prevalence of drug-induced nephropathy due to the great variety in its clinical and pathological presentations. Possible causes of AFR include decreased renal perfusion, damage to or vasoconstriction of renal blood vessels, injury to tubular cells, and allergic interstitial injury. Kidney failure is caused in part by the use of nephrotoxic drugs, which damage the kidneys' blood arteries, tubular cells, interstitial cells, and papillary structures. These negative effects are often the result of electrolyte imbalances, acid-base imbalances, abnormalities in the water balance, etc. These alterations may speed up the development of morbidities.
Servais H et al.,[20] Programmed cell death is a major contributor to renal toxicity and other kidney disorders in humans. Nephrotoxic drugs are common and cause kidney failure by activating the cell death pathway in the kidney's tubular cells. The processes that trigger apoptosis occur in two phases. The internal or intrinsic pathway involves the cell's organelles. The extrinsic route is an alternative method. The aforementioned mechanisms trigger the activation of two specific proteases, caspase-3 and caspase-7, which in turn leads to the development of characteristic morphologic markers of apoptosis, such as membrane blebbing, cell shrinkage, and DNA fragmentation.
Neesh Pannu et.al.,[15] The quantity of aminoglycosides in the tubular cells of the nephrons may disrupt normal cellular physiology and function. These accumulate in the lysosomal organelles of tubular cells. Protein synthesis and mitochondrial function, two typical cellular functions, are hampered. Changes like this may activate a pathway that leads to cell death. Gentamicin and other aminoglycoside antibiotics cause cell signalling and cell death by activating the calcium receptor found on tubular cells. Increased cytosolic calcium is a fascinating condition that has been associated to sepsis, immune-mediated disease, and glomerular illnesses.
Yaremi Quiros et.al.,[9] Epithelial cells that form the PCT have a brush border and are cuboidal in shape. These cells' apical surfaces also include many additional kinds of transporters, such as sodium/glucose symporters and sodium/proton antiporters. Tubular necrosis is caused by the antibiotic class known as aminoglycosides. Gentamicin accumulates in these cells when it is administered. Megalin and cubulin receptors are highly expressed in the cuboidal epithelial cells of the PCT. Gentamicin and other cation medications are transported into tubular cells through these receptors. Because of this, gentamicin accumulates. Additionally, these receptors are responsible for transporting certain protein molecules into the tubular cells. Molecules of gentamicin accumulate in the tubular cells of the PCT, namely in the endoplasmic reticulum and Golgi complexes. Subsequently, the accumulated molecule is concentrated in mitochondria and other cellular organelles, where it triggers the cell's intrinsic apoptosis process.
Ozaki N et.al., [24] Insight into the deleterious effects of aminoglycosides like gentamicin on tubular cells is provided. The antibiotic gentamicin has a role in the PCT cells' reprogrammed apoptosis as well. Due to gentamicin's damaging effects on the PCT cells' cellular components, the normal form of the cells is altered. Additionally, aminoglycoside drugs influence apical transporters.
Andrew Prayle et.al., [21] Renal tubular cells are stimulated to release cytochrome C by gentamicin, which in turn is stimulated by the mitochondria. This is the most crucial step in the apoptotic process. The resulting reactive oxidative species stimulate the development of stress-inducing genes such chaperones and oxidoreductases. Even when gentamicin fails to enter the cells, it might still have detrimental effects on the inside. Voltage-gated calcium channels on the apical surfaces of PCT cells open up in response to gentamicin molecules, enabling Ca2+ ions to enter the cells. This is due to the membrane Ca2+ receptor CaR.
Servais H et al., [28] While gentamicin serves a protective role inside lysosomes, it has nephrotoxic effects outside of these organelles. In response to caspase-3 activation, further steps include mitochondrial activation, cytochrome C release, and Bcl-2 expression. If gentamicin is found in the cytoplasm, it may have an effect on the mitochondria. Although polyunsaturated lipids may have a role, gentamicin has been shown to boost ROS generation in vitro.
III. METHODOLOGY
A. Study Design And Patients
Between July 2013 and June 2017, 11 tertiary medical centres in Switzerland, Germany, and Brazil participated in this international, multicenter, prospective study. As of the end of September, there were no available slots for follow-up visits in the first three months of 2017. A total of 156 individuals were diagnosed with central diabetic insipidus and/or hypotonic polyuria (16 and up).One of these conditions is present if the individual produces more than 50 ml of urine per kilogramme of body weight in a 24-hour period and the osmolality of the urine is less than 800 mOsm per kilogramme of body weight. Twelve persons were not able to take part in the trial due to exclusion criteria such as nephrogenic diabetic insipidus (which accounted for three of the dropouts). All participating institutions' institutional review boards have given their permission to the study's procedures. The parents or legal guardians of all minors who took part signed written permission forms. The only commercial support for this study came from Thermo Fisher Scientific, who funded the laboratory measurement of copeptin. All authors confirm that all necessary procedures were followed and that the data presented here are accurate and complete.
B. Procedures At Baseline
The water restriction test was conducted on a different day than the hypertonic saline infusion. Following the collection of pertinent medical background information, customary clinical and biochemical testing were performed. Although it was ultimately up to the discretion of the treating physician to order a head MRI, it was strongly recommended for all patients who had not had imaging during the prior three months prior to study enrollment. Each test required a 12-hour dry period before smoking or consuming alcohol, and a 24-hour break from diuretic or antidiuretic medication.
C. Test Protocols
- Indirect Water-Deprivation Test
For individuals with full diabetes insipidus or a high suspicion of it, the test's fluid restriction portion began at 6 a.m., whereas the water-deprivation portion began at midnight. We monitored blood pressure, heart rate, and weight every two hours, and collected urine to measure osmolality and volume. At 8 a.m., one hour before the desmopressin injection, blood was drawn (1 hour before the end of the test). The water deprivation test was stopped early if the patient had orthostatic hypotension (a rise in heart rate or a reduction in mean arterial blood pressure of more than 15%) or if their plasma sodium level rose beyond 150 mmol per litre.
2. Hypertonic Saline Infusion Test
Hypertonic saline was infused intravenously into the volunteers between 8 and 11 in the morning. 16 After a bolus infusion of 250 ml, a continuous infusion of 0.15 ml per kilogramme per minute of 3% saline is now taking place. Glucose, uric acid, osmolality, and sodium concentrations in the plasma were measured at 30-minute intervals. The salt levels in the vein blood were constantly checked to make sure they were higher than 150 mmol/L. Within 30 minutes after drawing blood for plasma copeptin measurement, patients drank 30 ml of water per kilogramme of body weight orally, followed by a 500 ml infusion of 5% glucose 40-60 minutes later.
D. Adverse Events And Symptom Burden
Patients used a 0-10 visual-analogue scale to estimate the severity of their symptoms in the clinical setting (very severe symptoms). We made sure to record any unfavourable results from experiments.
E. Test Interpretation And Preliminary Diagnosis
Patients were given a standard clinical practise based preliminary diagnostic and treatment plan before being discharged. Three months later, the patient had to return for a follow-up appointment so that the ultimate clinical result, treatment response, and accuracy of the original diagnosis could be assessed.
F. Diagnostic Criteria
- Indirect Water-Deprivation Test
According to the original and revised descriptions of the indirect water-deprivation test, a diagnosis of full central diabetes insipidus was made when desmopressin therapy resulted in a urine osmolality increase of more than 50 percent from a baseline value of less than 300 mOsm per kilogramme. Increases in urine osmolality of 300-800 mOsm per kilogramme caused by desmopressin were diagnostic of partial central diabetes insipidus. Primary polydipsia was diagnosed in patients whose peak urine osmolality was between 300 and 800 mOsm per kilogramme, and whose urine osmolality increased by less than 9 percent in response to desmopressin treatment.
2. Plasma Copeptin Stimulated by Water Deprivation
In order to improve the test's diagnostic accuracy in indirect water-deprivation investigations, researchers are keeping tabs on plasma copeptin levels before and after a morning desmopressin injection. A stimulated copeptin to plasma sodium ratio of 0.02 pmol/l or higher was shown to be diagnostic of primary polydipsia, whereas a baseline plasma copeptin level of less than 2.6 pmol/l was found to be diagnostic of complete central diabetes insipidus. Blood levels of the hormone copeptin increase after just 8 hours of dehydration (in picomoles per litre). Central diabetes insipidus is suspected when the level falls below 0.02 pmol/L.
3. Plasma Copeptin Stimulated by Hypertonic Saline Infusion
This research aimed to use copeptin stimulation by hypertonic saline as a diagnostic tool to distinguish between primary polydipsia and central diabetes insipidus. Copeptin levels below 4.9 pmol/l have been related to central diabetes insipidus, whereas copeptin levels over 4.9 pmol/l have been linked to primary polydipsia.
G. Final Reference Diagnosis
After reviewing each patient's medical history, clinical symptoms, water-deprivation test results, available laboratory and imaging data, and therapeutic response at the 3-month follow-up, two independent board-certified endocrinology experts blinded to the copeptin levels made the reference diagnoses. Diagnosis disagreements were resolved by the inclusion of a third expert's opinion in only 4% of the total 144 cases.
H. Laboratory Measurements
A plasma copeptin test was one of the procedures used to analyse the obtained blood samples (hematocrit, urine and plasma osmolality, and plasma sodium, creatinine, urea, potassium, calcium, glucose, albumin, and haemoglobin levels). Cerebral plasma copeptin concentrations were evaluated using a commercially available automated immunofluorescence assay.
I. Statistical Analysis
Differentiating primary polydipsia from central diabetes insipidus relied heavily on overall diagnostic accuracy, or the proportion of properly recognised individuals. All three cases had nephrogenic diabetic insipidus, but no other health issues were reported. characteristics of both the whole analytic sample and the subsample used in the methodology.
With this in mind, we set out to check whether (1) direct copeptin measurements taken during water deprivation were more accurate than indirect measurements, and (2) direct copeptin measurements taken during hypertonic saline infusion were comparable to those taken during water deprivation. To put it another way, the main hypothesis may be split up into smaller, testable hypotheses. It has been estimated that a research with 115 patients would have 90% power to demonstrate noninferiority of hypertonic saline-stimulated copeptin measurement versus water-deprivation-stimulated copeptin measurement, given that the diagnostic accuracy for water-deprivation-stimulated copeptin5 is 90%. By comparing the indirect water deprivation test, the copeptin response to hypertonic saline, and the copeptin reaction to water deprivation, one may identify primary polydipsia from partial central diabetes insipidus.
IV. RESULT
A. Baseline Characteristics
Sixty-two (42%) and eighty-two (58%) of the 141 patients (66%) who participated in the trial were diagnosed with central diabetes insipidus and primary polydipsia, respectively, at the conclusion of the 3-month follow-up period. From the 69 persons diagnosed with central diabetes insipidus, 36 (61%) had severe symptoms while the remaining 23 (23%), had just moderate symptoms (39 percent).
Some baseline features' existence varied considerably across the groups. The results of 97 people's brain MRIs were available. Primary polydipsia patients are more likely than central diabetes insipidus patients to have a hyperintense signal in the posterior region of T1-weighted imaging. Seventy percent of people with central diabetes insipidus do not have this signal, which is assumed to represent the pituitary arginine vasopressin level.
B. Primary Outcome
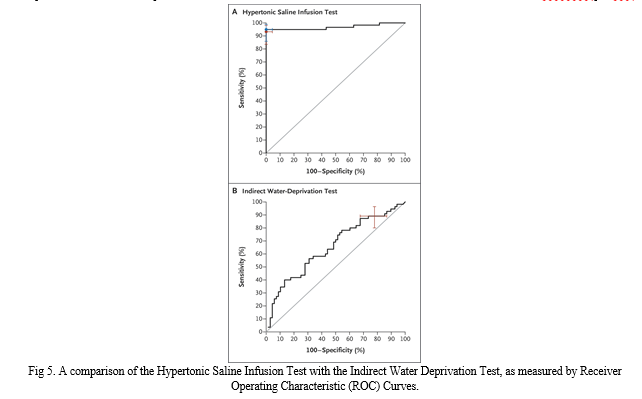
When compared to the alternative, hypertonic saline infusion has been found to be the most successful way of diagnosis (96.5 percent [95.1 to 98.6] vs. 76.6 percent [68.9 to 83.2]; P0.001). Comparison of individuals with primary polydipsia and partial central diabetes insipidus revealed that the hypertonic saline infusion test was much more accurate than the indirect water-deprivation test (95.2 percent [95 percent CI, 89.4 to 98.1] vs. 73.3 percent [95 percent CI, 63.9 to 81.2]; adjusted P 0.001).
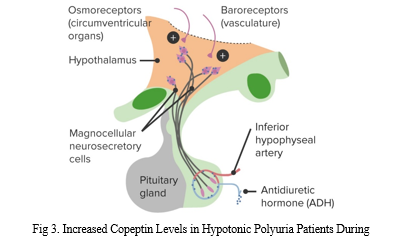
After measuring copeptin, infusing the patient with hypertonic saline improves the water-deprivation test's ability to distinguish between primary polydipsia and central diabetes insipidus (fig 3.). Hypertonic saline concentrations more than 4.9 pmol per litre had an area under the curve of 0.97, a sensitivity of 93.2% (95% CI, 83.5 to 98.2%), and a specificity of 100% for distinguishing between primary polydipsia and central diabetes insipidus (95 percent CI, 95.5 to 100.0). From 0.93 to 1.00, it was the range of outcomes (interquartile range [IQR]). At the best copeptin cutoff level (decided post hoc), 6.5 pmol per litre, diagnostic accuracy was 97.9% (95 percent CI, 93.9 to 99.6), sensitivity was 94.9 percent (95 percent CI, 85 percent to 98 percent), and specificity was 100%. Confidence Interval (95% CI): 95.5% - 100% .0]

Primary polydipsia may be diagnosed more reliably with indirect water deprivation testing than with the water deprivation-stimulated copeptin approach (44.0 percent [95 percent CI, 35.7 to 52.5] vs. 76.6 percent [95 percent CI, 68.9 to 83.2]). When diagnosing full-blown central diabetes insipidus, a copeptin level of less than 2.6 pmol per litre in the morning after overnight water restriction has a sensitivity of 78.4 percent. Confidence intervals (CIs) are a statistical method for expressing the range of possible outcomes of an event (70.6 to 84.9).
C. Secondary Outcomes And Burden Of Tests
There were no documented problems with the switch from water deprivation to hypertonic saline infusion in individuals who had both therapies (median score on the visual-analogue scale, 6 [interquartile range, 4 to 7] vs. 5 [interquartile range, 3 to 6]) Because of this, patient tolerability (how well a test goes for a patient) decreases (38 percent of patients preferred the water-deprivation test, whereas 62 percent preferred the hypertonic saline infusion test). Six subjects were diagnosed with primary polydipsia, five with central diabetes insipidus, and one with partial-onset central diabetes after having a hypertonic saline infusion (both patients had complete central diabetes insipidus). Twelve individuals had initial sodium concentrations between 140 and 144 millimoles per litre.
When water was severely limited, the number of adverse events rose to nine, and when hypertonic saline was administered, it rose to twenty-one. Desmopressin-induced hyponatremia was the most serious adverse event reported following the water-deprivation test.
Conclusion
Despite its rarity, DI may negatively affect a patient\'s quality of life if it is not properly managed. Getting the right diagnosis and starting treatment right away is crucial to improving the patient\'s quality of life. Central diabetic insipidus is brought on by a deficiency in ADH production from the posterior pituitary gland. As long as NDI is present, collecting duct tubular cells will not respond to the hypotensive hormone ADH. CDI, dipsogenic DI, and GDI may all result from an inadequate amount of ADH in the body. An unusually low osmotic thirst threshold causes dipsogenic DI by leading to excessive fluid consumption. Placental vasopressinase, an enzyme that breaks down the mother\'s adreno-sine dihydrocortisone, increases when GDI is present (ADH). Important methods for distinguishing between DI subtypes include measuring urine osmolality in reaction to water restriction, vasopressin response, and copeptin levels in response to osmotic stimulation. Copeptin and urine osmolality measurements may be useful in differentiating CDI from NDI when combined with water restriction and DDAVP treatment. Diagnostic magnetic resonance imaging (MRI) of the brain has shown some potential for use in establishing CDI. Dipsogenic DI may be identified in patients with a low osmotic thirst threshold and a very dilute urine. A GDI diagnosis may be made by measuring the urine and serum osmolality. The main goals of DI therapy are to improve patients\' quality of life and lessen the likelihood of complications due to fluid retention. To complement rehydration treatment, DDAVP is used to treat CDI. Despite popular belief, thiazide diuretics are effective in treating both CDI and NDI. As a first line of defence against NDI, reducing or avoiding exposure to the offending chemical is recommended. Once again, DDAVP does not influence either NDI or dipsogenic DI. When other treatments for dipsogenic DI have failed, antipsychotic medication may be used as a final option. To this day, DDAVP remains the medicine of choice for treating GDI. The appropriate treatment may make a huge difference in a patient\'s quality of life, so that\'s wonderful news for everyone.
References
[1] Maghnie M, Cosi G, Genovese E, Manca-Bitti ML, Cohen A, Zecca S, Tinelli C, Gallucci M, Bernasconi S, Boscherini B, Severi F, Arico M: Central diabetes insipidus in children and young adults. N Engl J Med 2000;343:998–1007. [2] Hensen J, Buchfelder M: The posterior pituitary and its disease; in Pinchera A, et al (eds): Endocrinology and Metabolism. New York, McGraw-Hill, 2001, pp 99–115. [3] Fujiwara TM, Bichet DG: Molecular biology of hereditary diabetes insipidus. J Am Soc Nephrol 2005;16:2836–2846. [4] Bichet DG: Vasopressin receptor mutations in nephrogenic diabetes insipidus. Semin Nephrol 2008;28:245–251. [5] Robinson A, Verbalis J: Posterior pituitary; in Kronenberg H, et al (eds): Williams Textbook of Endocrinology, ed 11. Philadephia, Saunders Elsevier, 2008, pp263–287. [6] Christensen JH, Rittig S: Familial neurohypophyseal diabetes insipidus – an update. Semin Nephrol 2006;26:209–223. [7] Barat C, Simpson L, Breslow E: Properties of human vasopressin precursor constructs: inefficient monomer folding in the absence of copeptin as a potential contributor to diabetes insipidus. Biochemistry 2004;43:8191–8203. [8] De Mota N, Reaux-Le Goazigo A, El Messari S, Chartrel N, Roesch D, Dujardin C, Kordon C, Vaudry H, Moos F, Llorens-Cortes C: Apelin, a potent diuretic neuropeptide counteracting vasopressin actions through inhibition of vasopressin neuron activity and vasopressin release. Proc Natl Acad Sci USA 2004;101:10464–10469. [9] Agre P: Nobel lecture. Aquaporin water channels. Biosci Rep 2004;24:127–163. [10] Engel A, Fujiyoshi Y, Agre P: The importance of aquaporin water channel protein structures. EMBO J 2000;19:800–806. [11] Robertson GL: Posterior pituitary; in Felig B, Frohman LA (eds): Endocrinology and Metabolism, ed 4. New York, McGraw Hill, 2001, p 234. [12] Braverman LE, Mancini JP, McGoldrick DM: Hereditary idiopathic diabetes insipidus: a case report with autopsy findings. Ann Intern Med 1965;63:503–508. [13] Ghirardello S, Garre ML, Rossi A, Maghnie M: The diagnosis of children with central diabetes insipidus. J Pediatr Endocrinol Metab 2007;20:359–375. [14] Willcutts MD, Felner E, White PC: Autosomal recessive familial neurohypophyseal diabetes insipidus with continued secretion of mutant weakly active vasopressin. Hum Mol Genet 1999;8:1303–1307. [15] Abu Libdeh A, Levy-Khademi F, Abdulhadi-Atwan M, Bosin E, Korner M, White PC, Zangen DH: Autosomal recessive familial neurohypophyseal diabetes insipidus: onset in early infancy. Eur J Endocrinol 2010;162:221–226. [16] Ye L, Li X, Chen Y, Sun H, Wang W, Su T, Jiang L, Cui B, Ning G: Autosomal dominant neurohypophyseal diabetes insipidus with linkage to chromosome 20p13 but without mutations in the AVP-NPII gene. J Clin Endocrinol Metab 2005;90:4388–4393. [17] Christensen JH, Siggaard C, Corydon TJ, Robertson GL, Gregersen N, Bolund L, Rittig S: Differential cellular handling of defective arginine vasopressin (AVP) prohormones in cells expressing mutations of the AVP gene associated with autosomal dominant and recessive familial neurohypophyseal diabetes insipidus. J Clin Endocrinol Metab 2004;89:4521–4531. [18] Davies J, Murphy D: Autophagy in hypothalamic neurones of rats expressing a familial neurohypophysial diabetes insipidus transgene. J Neuroendocrinol 2002;14:629–637. [19] Russell TA, Ito M, Yu RN, Martinson FA, Weiss J, Jameson JL: A murine model of autosomal dominant neurohypophyseal diabetes insipidus reveals progressive loss of vasopressin-producing neurons. J Clin Invest 2003;112:1697–1706. [20] Wahlstrom JT, Fowler MJ, Nicholson WE, Kovacs WJ: A novel mutation in the preprovasopressin gene identified in a kindred with autosomal dominant neurohypophyseal diabetes insipidus. J Clin Endocrinol Metab 2004;89:1963–1968. [21] Christensen JH, Siggaard C, Corydon TJ, Robertson GL, Gregersen N, Bolund L, Rittig S: Impaired trafficking of mutated AVP prohormone in cells expressing rare disease genes causing autosomal dominant familial neurohypophyseal diabetes insipidus. Clin Endocrinol 2004;60:125–136. [22] Ito M, Yu RN, Jameson JL: Mutant vasopressin precursors that cause autosomal dominant neurohypophyseal diabetes insipidus retain dimerization and impair the secretion of wild-type proteins. J Biol Chem 1999;274:9029–9037. [23] Friberg MA, Spiess M, Rutishauser J: Degradation of wild-type vasopressin precursor and pathogenic mutants by the proteasome. J Biol Chem 2004;279:19441–19447. [24] Maghnie M, Ghirardello S, De Bellis A, di Iorgi N, Ambrosini L, Secco A, De Amici M, Tinelli C, Bellastella A, Lorini R: Idiopathic central diabetes insipidus in children and young adults is commonly associated with vasopressin-cell antibodies and markers of autoimmunity. Clin Endocrinol 2006;65:470–478. [25] Pivonello R, De Bellis A, Faggiano A, Di Salle F, Petretta M, Di Somma C, Perrino S, Altucci P, Bizzarro A, Bellastella A, Lombardi G, Colao A: Central diabetes insipidus and autoimmunity: relationship between the occurrence of antibodies to arginine vasopressin-secreting cells and clinical, immunological, and radiological features in a large cohort of patients with central diabetes insipidus of known and unknown etiology. J Clin Endocrinol Metab 2003;88:1629–1636. [26] Bellastella G, Rotondi M, Pane E, Dello Iacovo A, Pirali B, Dalla Mora L, Falorni A, Sinisi AA, Bizzarro A, Colao A, Chiovato L, De Bellis A: Predictive role of the immunostaining pattern of immunofluorescence and the titers of antipituitary antibodies at presentation for the occurrence of autoimmune hypopituitarism in patients with autoimmune polyendocrine syndromes over a five-year follow-up. J Clin Endocrinol Metab 2010;95:3750–3757. [27] Maghnie M, Genovese E, Sommaruga MG, Arico M, Locatelli D, Arbustini E, Pezzotta S, Severi F: Evolution of childhood central diabetes insipidus into panhypopituitarism with a large hypothalamic mass: is ‘lymphocytic infundibuloneurohypophysitis’ in children a different entity? Eur J Endocrinol 1998;139:635–640. [28] De Jersey J, Carmignac D, Le Tissier P, Barthlott T, Robinson I, Stockinger B: Factors affecting the susceptibility of the mouse pituitary gland to CD8 T-cell-mediated autoimmunity. Immunology 2004;111:254–261. [29] Tzou SC, Lupi I, Landek M, Gutenberg A, Tzou YM, Kimura H, Pinna G, Rose NR, Caturegli P: Autoimmune hypophysitis of SJL mice: clinical insights from a new animal model. Endocrinology 2008;149:3461–3469. [30] Imura H, Nakao K, Shimatsu A, Ogawa Y, Sando T, Fujisawa I, Yamabe H: Lymphocytic infundibuloneurohypophysitis as a cause of central diabetes insipidus. N Engl J Med 1993;329:683–689. [31] Mirocha S, Elagin RB, Salamat S, Jaume JC: T regulatory cells distinguish two types of primary hypophysitis. Clin Exp Immunol 2009;155:403–411.
Copyright
Copyright © 2023 Aashi Singh Bhadouria, Ishan Singh Bhadouria. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET54580
Publish Date : 2023-07-02
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online