

Ijraset Journal For Research in Applied Science and Engineering Technology
Drug Response Prediction Using XGBOOST
Authors: P. Senthil, Princy Joseph, B. Praveen Kumar, B. Naresh, V. Vamshi
DOI Link: https://doi.org/10.22214/ijraset.2024.59337
Certificate: View Certificate
Abstract
An essential issue in computational personalised medicine is the prediction of drug responses. There have been several proposals for approaches to this problem that rely on machine learning, particularly deep learning. Nevertheless, these approaches often portray the medications as strings, an implausible representation of molecules. Furthermore, there has been a lack of comprehensive consideration of interpretation, such as whether mutations or copy number aberrations contribute to the medication response. Graph DRP, a new approach based on graph convolution networks, is suggested as a solution to the issue in this research. Cell lines were displayed as double vectors of genetic abnormalities in Graph DRP, whereas medications were shown as sub-atomic charts that straightforwardly caught the bonds among particles.
Introduction
I. INTRODUCTION
One idea behind personalised medicine is to use the correct medication at the appropriate time in the right amount. Thus, it is crucial in biomedical research to estimate the pharmacokinetic response of each individual patient using their unique biological features (e.g., omics data). Nevertheless, there is a lack of quality and quantity of standardised data about patients' treatment responses. When it comes to TCGA data, there has been very little research on medication response for cancer patients [1]. As a result, doing extensive studies on this area has become more hard. Luckily, computational approaches for drug response prediction have been developed thanks to large-scale programmes like GDSC, CCLE, and NCI60 that study drug response in "artificial patients" (i.e., cell lines).
The DREAM challenge for drug responsiveness expectation was truly begun, and a few examination gatherings have put up approaches for it . When it comes to data and model integration, most of these approaches are machine learning oriented. To combine different kinds of cell line - omics data with response data, for instance, multiple-kernel and multiple-task learning methods were suggested. In addition, several models were integrated using ensemble learning methodologies . Similarly, network-based approaches have been suggested that use similarity networks (such as those involving structural similarities between medications or biological likenesses between cell lines) and known responses from drug cell lines.
Also, drug response prediction has made use of gene regulatory networks and protein interaction. Since AI based techniques have demonstrated compelling in information and model joining, drug reaction expectation has by and large been drawn nearer methodically. Predefined characteristics, such as drug structural properties and cell line -omics profiles, are often used to describe medications and cell lines alike. A variety of classic AI based calculations frequently experience the "little n, huge p"issue since there are fewer cell lines than qualities in - omics profiles of cell lines. Thus, regular AI based algorithms can only go so far in terms of prediction accuracy.
II. RELATED WORK
In the quest for innovation and efficiency, modern projects frequently rely on existing solutions as fundamental building blocks for development. This approach not only recognizes the expertise and advancements of those who came before us but also nurtures a collaborative ecosystem where ideas can evolve and confront new challenges. In our project, we wholeheartedly embrace this ethos, conscientiously integrating elements from existing solutions to enrich our endeavor. These existing solutions serve as guiding lights, offering insights and frameworks that shape the direction of our project.
A. Graph Convolutional Network for Drug Response Prediction (GRAPHDRP)
The proposed show of sedate reaction forecast is appeared in Fig 1. The input information incorporates chemical data of drugs and genomic highlights of cell lines counting changes and duplicate number variations (i.e., genomic abnormality).For the sedate highlights, the drugs spoken to in Grinsorganize were downloaded from Pub Chem. At that point, RD Kit, an open-source chemical informatics program was utilized to build a atomic chart reflecting connect-activities between the iotas interior the sedate.
Iota highlight plan from Deep Chem was utilized to portray a hub within the chart. Each hub contains five sorts of particle features: particle image, particle degree calculated by the number of bonded neighbors and Hydrogen, the whole number of Hydrogen, verifiab le esteem of the molecule, and whether the iota is fragrant.
These iota highlights constituted a multi-dimensional twofold include vector. On the off chance that there exists a bond among a match of particles, an edge is set. As a result, an circuitous, parallel chart with ascribed hubs was built for each input Grins string. A few chart convolutional arrange models, counting GCN, GAT, GIN and combined GAT-GCN design, were utilized to learn the highlights of drugs. We utilized the same approach as other models since 1D convolution with a huge part has the capacity to combine genomic truncation within thegenomic highlights to create great expectations. In addition, 1D pooling was too utilized to decrease the measure of input feature at that point 1D convolutions can learn unique highlights from genomic highlights. The genomic highlights of cell lines were spoken to in one-hot encoding. 1D Convolutional neural organize (CNN) layers were utilized to memorize idle highlights on those information. At that point the yield was smoothed to 128 measurement vector of cell line representation.
B. Graph Convolutional Networks (GCN)
Formally, a chart for a given medicate G = (V, E) was put away within the frame of two networks, counting include framework X and contiguousness framework A. X ∈ RN×F comprises of N hubs in the chart and each hub is spoken to by F-dimensional vector. A ∈ RN×N shows the edge connection between hubs. The initial chart convolutional layer takes two lattices as input and points to deliver a node-level yield with C highlights each hub. The layer is characterized as where W ∈ RF ×C is the trainable parameter lattice.In any case, there are two primary downsides. To begin with, for each hub, all include vectors of all neighboring hubs were summed up but not the hub itself. Moment, framework A was not normalized, so the duplication with A will alter the scale of the highlight vector. GCN show was presented to unravel these impediments by including personality network to A and normalizing A.
C. Graph Attention Networks (GAT)
Self-attention technique has been shown to be self-sufficient for state-of-the-art-level results on machine translation Inspired by this idea, we used self-attention technique in graph convolutional network in GAT. We adopted a graph attention network (GAT) in our model. The proposedGAT architecture was built by stacking a graph attention layer. The GAT layer took the node feature vector x, as input then applied a linear Transformation to every node by a weight matrix W. Then the attention coefficients is computed at every pair of nodes that the edge exists.
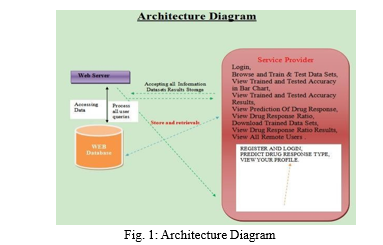
III. METHODS AND EXPERIMENTAL DETAILS PROPOSED METHODS
A. Decision Tree Classifiers
Effectiveness has been achieved using choice tree classifiers in a wide assortment of spaces. Ready to separate engaging dynamic data from gave information is their most notable quality. A decision tree may be constructed using a collection of training data. The following is the technique for such age utilizing the arrangement of articles (S), where every thing has a place with one of the classes C1, C2,..., Ck
B. Gradient Boosting
As a machine learning approach, gradient boosting has many applications, including classification and regression. Ensembles of weak prediction models, most often decision trees, are what it uses to generate a prediction model. One and two A technique known as gradient-boosted trees is produced at the point when a choice tree is utilized as the powerless student. As a rule, this approach accomplishes improved results than irregular forest. The development of a slope supported trees model follows similar stage-wise example as past helping methods; notwithstanding, it develops these methodologies by empowering the enhancement of any differentiable misfortune capability.
IV. LOGISTIC REGRESSION CLASSIFIERS
In logistic regression, a group of independent variables is utilized to concentrate on the connection between a clear cut subordinate variable and those factors. When the dependant variable may only take on two values—for example, yes or no—the method is known as logistic regression. When the dependent variable, such "married," "single," "divorced," or "widowed," may take on three or more distinct values, multinomial logistic regression is often used. When it comes to analysing variables having a categorical answer, strategic relapse is in direct rivalry with discriminant examination.
A. Naïve Bayes
One supervised learning technique that relies on an oversimplified premise is the naive bayes approach. This method presumes that the existence or absence of one class characteristic has no relation to the existence or absence of any other feature. This being said, it still seems to be efficient and durable. Other supervised learning approaches can't match its performance. The literature has put out a number of explanations for this. Our focus in this session is on an explanation that relies on representation bias. The naive bayes classifier, like linear discriminant analysis, logistic regression, and linear support vector machines, is a linear classifier.
B. Random Forest
A troupe learning method for order, relapse, and different issues, irregular timberlands (now and then called arbitrary choice woodlands) work by building an enormous number of choice trees during preparing. While doing a characterization challenge, the irregular woods will give the class that most of trees have picked. The normal or mean expectation from each tree is offered back for relapse errands. In 1995, Tin Kam Ho[1] imagined the principal arbitrary choice timberland calculation by utilizing the irregular subspace strategy. This strategy, as indicated by Ho's portrayal, is a method for putting Eugene Kleinberg's "stochastic segregation" way to deal with order into practice.
C. SVM
The goal of discriminant machine learning in classification problems is to derive a discriminant capability that can precisely anticipate names for recently obtained occasions utilizing an id (free and indistinguishably disseminated) preparing dataset. A discriminant characterization capability might take an information point x and spot it into one of the classes engaged with the grouping position, rather than generative AI methods that need computations of restrictive likelihood disseminations. By analytically solving the convex optimization issue, support vector machines (SVMs) consistently produce the same optimum hyperplane value, setting them apart from perceptrons and genetic algorithms (GAs), two of the most popular classification techniques in machine learning.
|
MODEL TYPE |
ACCURACY |
|
NAIVE BAYES |
90.15748314 |
|
SVM |
92.25721784 |
|
LOGISTIC REGRESSION |
91.11986001 |
|
DECISION TREE CLASSIFIER |
88.62682169 |
|
KNEIGHBOUR CLASSIFIER |
82.72090988 |
|
XGB CLASSIFIER |
87.75153105 |
Table : Metrics
V. IMPLEMENTATION
Using XGBoost for drug response prediction is a common and effective approach. You can utilize features like gene expression levels, genomic data, and other relevant molecular information as input for your model. Ensure proper data preprocessing, feature engineering, and model tuning for optimal performance. Using XGBoost for drug response prediction is a common and effective approach. You can utilize features like gene expression levels, genomic data, and other relevant molecular information as input for your model. Ensure proper data preprocessing, feature engineering, and model tuning for optimal performance.
- Data Collection and Exploration: Gather data on drug responses, considering factors like cell lines or patients and their corresponding responses to different drugs. Explore the dataset to understand its characteristics, identify missing values, and gain insights into potential features.
- Data Preprocessing: Clean the data by handling missing values and outliers. Encode categorical variables and standardize/normalize numerical features. Split the data into training and testing sets.
- Model Selection: Choose XGBoost as your predictive model due to its ability to handle complex relationships in data and manage high-dimensional feature spaces. Define your target variable (e.g., drug response) and train the model on the training dataset.
- Hyper parameter Tuning: Fine-tune XGBoost hyperparameters through techniques like grid search or random search to optimize the model's performance. Adjust parameters such as learning rate, tree depth, and regularization to avoid overfitting.
- Training the Model: Train the XGBoost model on the training set, allowing it to learn the patterns and relationships within the data.
- Evaluation: Evaluate the model on the testing set using metrics like accuracy, precision, recall, or area under the ROC curve (AUC-ROC), depending on the nature of your prediction problem.
- Interpretability: Analyze feature importance to understand which molecular features contribute significantly to the drug response prediction.
- Deployment: Once satisfied with the model's performance, deploy it for making predictions on new, unseen data. Remember to iterate on these steps as needed, and continually refine your model based on new data or insights gained from its performance.
A. Interfaces


Conclusion
Our work introduced Graph DRP, a new approach to drug response prediction. Instead of using strings to represent drug molecules, our model used graphs, and cellines were recorded using one-hot vector design. Then, at that point, 1D convolutional layers were utilized to gain proficiency with the cell-line portrayal, and diagram convolutional layers were used to learn the compound features. We then utilised the drug and cell-line representations together to forecast the IC50 value. This study employed four different graph neural network (GCN, GAT, GIN, and a mix of GAT and GCN) types to learn pharmacological characteristics. The state-of-the-art technique, TCNNS, used SMILES strings to represent drug compounds, and we compared our method to it. We discovered that some cancers are sensitive to the IC50 values of Bortezomib and Epothilone B, and we also determined that these medications had the lowest IC50 values.
References
[1] Lavecchia, “Deep learning in drug discovery: opportunities, challenges and future prospects,” Drug Discovery Today, 2019. [2] Karimi, D. Wu, Z. Wang, and Y. Shen, “DeepAffinity: interpretable deep learning of compound–protein affinity through unified recurrent and convolutional neural networks,” Bioinformatics, vol. 35, no. 18, pp. 3329–3338, 2019. [3] Tan, O. F. O¨ zgu¨ l, B. Bardak, I. Eks¸iog? lu, and S. Sabuncuoglu, “Drug response prediction by ensemble learning and drug-induced gene expression signatures,” Genomics, vol. 111, no. 5, pp. 1078–1088, 2019. [4] Gonczarek, J. M. Tomczak, S. Zareba, J. Kaczmar, P. Dabrowski, and M. J. Walczak, “Interaction prediction in structure-based virtual screening using deep learning,” Computers in Biology and Medicine, vol. 100, pp. 253–258, 2018. [5] O¨ ztu¨ rk, A. O¨ zgu¨ r, and E. Ozkirimli, “DeepDTA: deep drug– target binding affinity prediction,” Bioinformatics, vol. 34, no. 17, pp. i821–i829, 2018. [6] T. Nguyen and D.-H. Le, “A matrix completion method for drug response prediction in personalized medicine,” in Proceedings of the International Symposium on Information and Communication Technology, 2018, pp. 410–415. [7] H. Le and V.-H. Pham, “Drug response prediction by globally capturing drug and cell line information in a heterogeneous network,” Journal of Molecular Biology, vol. 430, no. 18, pp. 2993–3004, 2018. [8] H. Le and D. Nguyen-Ngoc, “Multi-task regression learning for prediction of response against a panel of anti-cancer drugs in personalized medicine,” in Proceedings of the International Conference on Multimedia Analysis and Pattern Recognition (MAPR). IEEE, 2018, pp. 1–5. [12] K. Matlock, C. De Niz, R. Rahman, S. Ghosh, and R. Pal, “Investigation of model stacking for drug sensitivity prediction,” BMC Bioinformatics, vol. 19, no. 3, p. 71, 2018. [9] Turki and Z. Wei, “A link prediction approach to cancer drug sensitivity prediction,” BMC Systems Biology, vol. 11, no. 5, p. 94, 2017. [10] Azuaje, “Computational models for predicting drug responses in cancer research,” Briefings in Bioinformatics, vol. 18, no. 5, pp. 820–829, 2017. [11] I. I. Baskin, D. Winkler, and I. V. Tetko, “A renaissance of neural networks in drug discovery,” Expert Opinion on Drug Discovery, vol. 11, no. 8, pp. 785–795, 2016. [12] C. Pereira, E. R. Caffarena, and C. N. dos Santos, “Boosting docking-based virtual screening with deep learning,” Journal of Chemical Information and Modeling, vol. 56, no. 12, pp. 2495–2506, 2016. a. Zhang, H.Wang, Y. Fang, J.Wang, X. Zheng, and X. S. Liu, “Predicting anticancer drug responses using a dual-layer integrated cell line-drug network model,” PLoS Computational Biology, vol. 11,no. 9, 2015. b. Wan and R. Pal, “An ensemble based top performing approach for NCI-DREAM drug sensitivity prediction challenge,” PLoS ONE, vol. 9, no. 6, 2014. c. S. Jang, E. C. Neto, J. Guinney, S. H. Friend, and A. A. Margolin, “Systematic assessment of analytical methods for drug sensitivity prediction from cancer cell line data,” in Biocomputing. World Scientific, 2014, pp. 63–74. d. C. Costello, L. M. Heiser, E. Georgii, M. G¨onen, M. P. Menden, N. J. Wang, M. Bansal, P. Hintsanen, S. A. Khan, J.-P. Mpindi et al., “A community effort to assess and improve drug sensitivity prediction algorithms,” Nature Biotechnology, vol. 32, no. 12, p. 1202, 2014. e. G¨onen and A. A. Margolin, “Drug susceptibility prediction against a panel of drugs using kernelized Bayesian multitask learning,” Bioinformatics, vol. 30, no. 17, pp. i556–i563, 2014. f. N. Weinstein, E. A. Collisson, G. B. Mills, K. R. M. Shaw, B. A. Ozenberger, K. Ellrott, I. Shmulevich, C. Sander, J. M. Stuart, C. G. A. R. Network et al., “The Cancer Genome Atlas Pan-Cancer analysis project,” Nature Genetics, vol. 45, no. 10, p. 1113, 2013. g. Yang, J. Soares, P. Greninger, E. J. Edelman, H. Lightfoot, S. Forbes, N. Bindal, D. Beare, J. A. Smith, I. R. Thompson et al., “Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells,” Nucleic Acids Research, vol. 41, no. D1, pp. D955–D961, 2012. h. Barretina, G. Caponigro, N. Stransky, K. Venkatesan, A. A. Margolin, S. Kim, C. J. Wilson, J. Leh´ar, G. V. Kryukov, D. Sonkin et al., “The Cancer Cell Line Encyclopedia enables predictive modeling of anticancer drug sensitivity,” Nature, vol. 483, no. 7391, pp. 603– 607, 2012.
Copyright
Copyright © 2024 P. Senthil, Princy Joseph, B. Praveen Kumar, B. Naresh, V. Vamshi. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET59337
Publish Date : 2024-03-23
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online