

Ijraset Journal For Research in Applied Science and Engineering Technology
Extensive Review Paper on Computational Catalyst Discovery using Machine Learning Algorithms
Authors: Vinod Ingale, Saniya Devale, Anushka Khedekar, Manasi Gaikwad, Dnyaneshwari More
DOI Link: https://doi.org/10.22214/ijraset.2024.65221
Certificate: View Certificate
Abstract
As the world continues to grapple with energy scarcity and climate change, the future of our energy supply faces increasing challenges. These technologies offer the opportunity to develop efficient, carbon-neutral ways to store and produce energy. But doing so requires discovering efficient and economical materials that can speed up chemical processes. One way to find effective catalysts is to use molecular experiments. Specifically, each simulation simulates the interaction of the catalyst surface with molecules typically found in electrochemical reactions. By accurately predicting these interactions, the effect of the catalyst on the overall rate of the chemical reaction can be predicted.
Introduction
I. INTRODUCTION
Machine learning evolved from a purely theoretical field to one of the most influential technologies in modern science. While initially focused on elementary statistical models, further development was triggered by improvements in computational resources toward more advanced algorithms such as neural networks and layered deep learning architectures. By the early 2000s, machine learning had already established its significant foothold in the fields of image recognition and natural language processing. However, with increasing data availability and the advancement of computational resources, the machine-learning paradigm tackled even more complex domains such as chemistry and materials science. Today, such a platform is becoming essential in solving problems previously considered too challenging for conventional approaches.
Concurrently, catalysis has been a long-standing backbone of chemical processes, granting the facilitation of innumerable reactions essential to many sectors, ranging from energy to pharmaceuticals. Noteworthy, catalyst discovery and optimization were historically slow and labour-intensive methods that depended on the trial-and-error model. It implied synthesizing and testing a great number of materials in search of the ideal combination with the requisite ability to promote a given reaction. Consequently, with the rising demand for more efficient catalysts envisioned within renewal energy generation, features of traditional experimental procedures themselves came to the fore. Beginning in the early years of the 21st century, many computational methods started to alter the landscape of catalyst research. Scientists began using DFT to conduct atomic-level simulations of chemical reactions, predicting catalyst behaviour in advance of an experiment. Consequently, this reduced catalyst discovery time and cost. While DFT proved to be a significant step forward, it still proved to be expensive and limited in execution regarding complex systems and large datasets. With the desire to cover more complicated reactions for renewable energy, the restrictions of DFT began to show.
The combination of machine learning and catalysis offered a promising solution to these challenges. ML models, trained on large datasets, can predict reaction outcomes and material properties with remarkable accuracy. For example, through such a layer of pattern-building, ML, at least in some part, gets around the exhaustive need to carry out physical experiments, thereby accelerating catalyst discovery. Recently, there has been an increased interest in the application of ML for improving catalyst design, especially in renewable energy, for where the need for solutions has efficiently scalable needs. By predicting how materials behave as catalysts would speed up the development of new technologies through ML. Recalling the OC20 dataset in 2020, one of the amounts of major milestone moments in the field, this was the first time it had become a reality. With over 1.2 million DFT relaxations of molecular adsorptions on metal surfaces, it availed unparalleled resources for the trainings of ML in catalysis. The dataset contains adsorbates that are relevant to renewable energy-water-splitting and solar-fuel-synthesizing reactions-redox. The sparing of this data provided ample material for the development of very advanced types of ML models, particularly Graph Neural Networks (GNNs), which are able to model complex catalytic systems with great accuracy. However, the OC20's main focus was metal catalysts, not oxides, which is critical for many energy storage and conversion phenomena. Benefit to answer this shortcoming was provided by the release of the OC22 dataset focused on oxide surfaces. These are harder to model due to their structural complexity, yet they do play important roles in certain renewable energy reactions. The OC22 dataset was built up to train ML models predicting oxide catalysts' activity and stability, with nearly 10 million individual calculations.
It is through the connection of the science of catalysis and machine learning that researchers can, at a really dimension, explore through a wide range of catalytic materials and reactions. With that integration leading to more discoveries at faster rates and reactions that are more efficient, catalyst research is, on the whole, set to bring about solutions involving clean energy. With future improvements in machine learning models and a continued expansion based on datasets like OC20 and OC22, the future of catalyst research opens up new horizons in attempts to assist humanity solve some of its pressing energy problems.
II. LITERATURE SURVEY
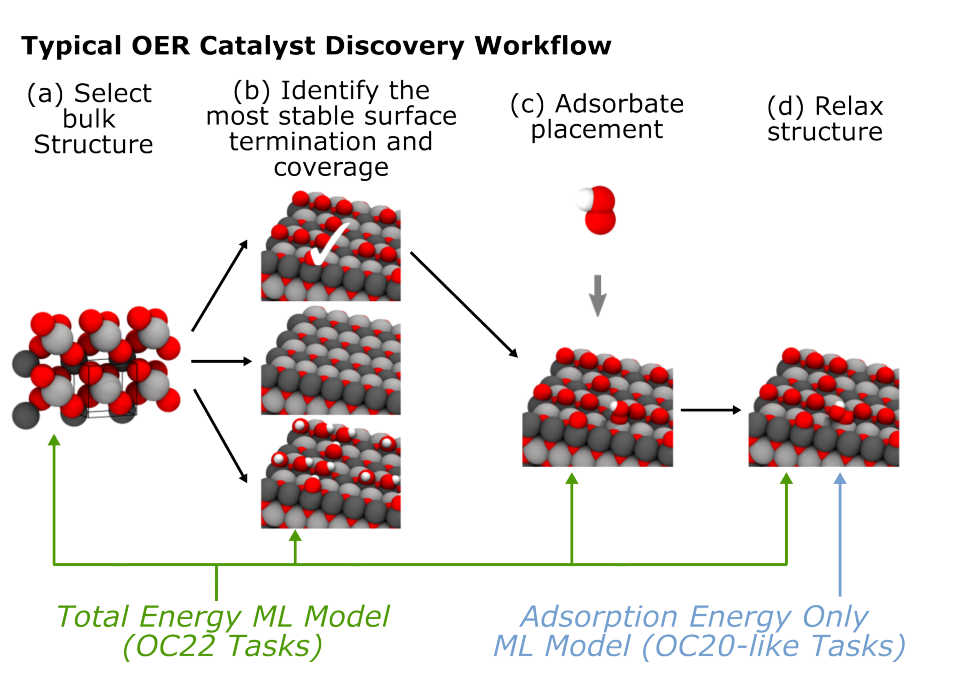
Fig 1: Catalyst Discovery Workflow (Tran et al., 2022[1])
A. Bulk Selection
The study focuses on a dataset of 4,728 unary (AxOy) and binary (AxByOz) metal-oxide materials sourced from the Materials Project. This dataset includes various metal and semi-metal combinations, particularly featuring cerium (Ce) and lutetium (Lu) due to their catalytic properties. The criteria for selection prioritized chemical diversity, focusing on the five lowest energy bulk materials above the hull with fewer than 150 atoms, despite some having energies exceeding 0.1 eV/atom. The materials were categorized based on their electronic band gaps into metallic, semiconducting, and insulating types, reflecting their potential for photocatalytic applications. The analysis also included 173 rutile structures. While the selection favoured chemical diversity, it acknowledged that many chosen materials might lack electrochemical stability, which is crucial for electrocatalytic applications. Previous Pourbaix analyses highlighted that only 26 of the 51 elements considered are stable in aqueous environments.
B. Surface Selection
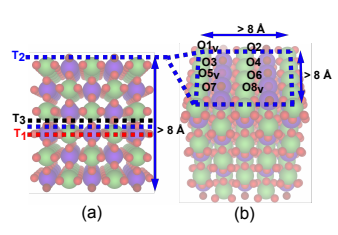
Fig 2: Surface Selection (Tran et al., 2022[1]).
The dataset was constructed by randomly sampling 4,286 bulk oxides from an original set of 4,728, focusing on slabs with fewer than 250 atoms. Each slab was created by exploring all possible surface terminations with a maximum Miller index of 3. A randomly chosen termination was replicated to a depth of at least 8 Å and a width of at least 8 Å in each cross-sectional direction.
The surface of each slab was then modified by adding a random number of oxygen vacancies, which serve as active sites for reactions like CO2 capture and the oxygen evolution reaction (OER). Oxygen lattice sites on the surface were identified, and a random number of surface oxygen were removed symmetrically from both surfaces to prevent non-physical dipole moments that could affect DFT energy calculations.
C. Initial Structure Generation
In constructing adsorbate+slab models, a random adsorbate is selected from a set that includes intermediates of the Oxygen Evolution Reaction (OER) (e.g., O*, OH*, H2O*, OOH*, O2*) as well as monatomic H*, N*, C*, and CO*. Adsorbates bind to surface oxygen, under-coordinated surface metal, or oxygen vacancies, with multiple adsorbates allowed, but they are separated to avoid overcrowding. Adsorbates are placed on surface sites with strategies ensuring proper binding, with oxygen-containing adsorbates binding to metal atoms. Adsorbate molecules may also form new molecules via surface oxygen binding, enabling the exploration of intermediate surface reactions. Lastly, adsorbates are allowed rotational freedom about the surface normal.
D. Structure Relaxation
System DFT energies were referenced to represent adsorption energies. Adsorption energies were calculated according to the Equation given, where Esys is the DFT energy of the combined surface (i.e. slab) and adsorbate — this energy can be from both relaxed and intermediate structures. The reference energies for each system, Eslab and Egas are the DFT energy of the relaxed surface and adsorbate molecule respectively. The value of Egas for each adsorbate was computed as a linear combination of N2, H2O, CO, and H2 resulting in the atomic energies found in the supplementary.
Ead = Esys–Eslab–Egas (1)
The OC22 dataset employs different computational settings compared to OC20, using the Perdew-Burke-Ernzerhof (PBE) functional within the generalized gradient approximation (GGA), which is more suitable for modeling surface reactions on oxides. OC22 also includes the Hubbard U correction for transition metal oxides, following recommendations from the Materials Project, to account for strong electron correlations. In contrast to OC20, all OC22 calculations are spin-polarized to handle the magnetic states of metal oxides, with only one magnetic polymorph studied per material.
A key difference in OC22 is the full relaxation of both the slab and adsorbate+slab systems, leading to more accurate surface energy calculations. This approach contrasts with OC20, where only adsorbates and surface atoms were relaxed. The dataset allows for exploration beyond adsorption energies, enabling studies of complete reaction mechanisms on metal oxides.
III. TASKS
A. S2EF (Structure to Energy and Forces)
Objective: Predict the total energy and atomic forces of a material based on its atomic structure.
Use Case: S2EF is a detailed method that not only predicts the energy of a material's atomic configuration but also provides forces, which are crucial for relaxing structures and understanding atomic dynamics.
B. IS2RS (Initial Structure to Relaxed Structure)
Objective: Predict the final (relaxed) atomic structure based on an initial structure.
Use Case: IS2RS is used when understanding the geometric arrangement of atoms after relaxation is important, such as in studying surface reconstructions or adsorption in catalysis.
C. IS2RE (Initial Structure to Relaxed Energy)
Objective: Predict the relaxed energy of a material given its initial atomic structure.
Use Case: IS2RE is particularly useful for quickly assessing stability without the computational cost of predicting forces or detailed atomic rearrangements.
IV. IS2RE DATASET USAGE
In this project, the IS2RE (Initial Structure to Relaxed Energy) approach has been selected over S2EF and IS2RS due to the following reasons:
A. Computational Efficiency
- IS2RE directly predicts the relaxed energy of a material based on its initial structure without needing to compute intermediate forces or run full structural relaxations. This makes it significantly faster and computationally less expensive compared to S2EF, which requires calculating atomic forces in addition to energy.
- While IS2RS provides detailed insights into the final atomic arrangement, it requires the model to infer atomic positions during relaxation. In contrast, IS2RE skips this step, offering a more streamlined and quicker evaluation of material stability, which is crucial in projects where evaluating a large number of candidate materials is necessary.
B. Focus on Material Stability
- The core goal of this project is to evaluate the stability of various catalyst materials, which is best represented by the final relaxed energy. IS2RE provides a direct prediction of this stability metric, making it the ideal choice for screening materials.
- S2EF offers additional information, such as forces and structural dynamics, but this level of detail is not necessary for this project, where the overall energy stability is the primary concern. Similarly, IS2RS offers insights into the final structure, but this is not as critical as knowing whether the material is thermodynamically stable.
C. Simplified Evaluation Process
- IS2RE simplifies the evaluation process by focusing solely on predicting energy, making it easier to interpret and use for decision-making in the context of catalyst design. The output—a single energy value—directly reflects the stability of the material, allowing for easy comparison between different candidates.
- In contrast, S2EF provides both energies and forces, which would require additional steps (e.g., force-driven relaxations) to fully understand the stability of a material. IS2RS involves interpreting final atomic positions, which adds complexity when the primary goal is to rank materials based on energy.
D. Applicability to Large Datasets
Given the large number of materials being studied in this project, IS2RE is particularly well-suited to handle high-throughput screening. Predicting relaxed energies for a wide array of materials allows for rapid identification of promising candidates, while more complex models like S2EF would require significantly more computational resources to achieve similar results.
V. GRAPH NEURAL NETWORK FOR MATERIAL STABILITY:
The machine learning model used in IS2RE is typically a graph neural network (GNN) or a variant of a message-passing neural network (MPNN). These types of models are well-suited for processing atomic structures, which can be represented as graphs where atoms are the nodes and bonds (or interactions) between atoms are the edges.
Here is how the model works:
A. Graph Representation
The atomic structure of a material is represented as a graph. Each atom is a node, and bonds (or interactions) between atoms are the edges. The model can capture both the types of atoms (node features) and their interactions (edge features).
B. Message Passing
The GNN/MPNN processes the graph by iteratively updating the information at each node (atom) based on its neighbours. This allows the model to capture the local environment of each atom, which is crucial for predicting properties like energy.
C. Prediction Layer
After several rounds of message passing, the model aggregates the information from all atoms in the structure and outputs a prediction of the relaxed energy for the entire material.
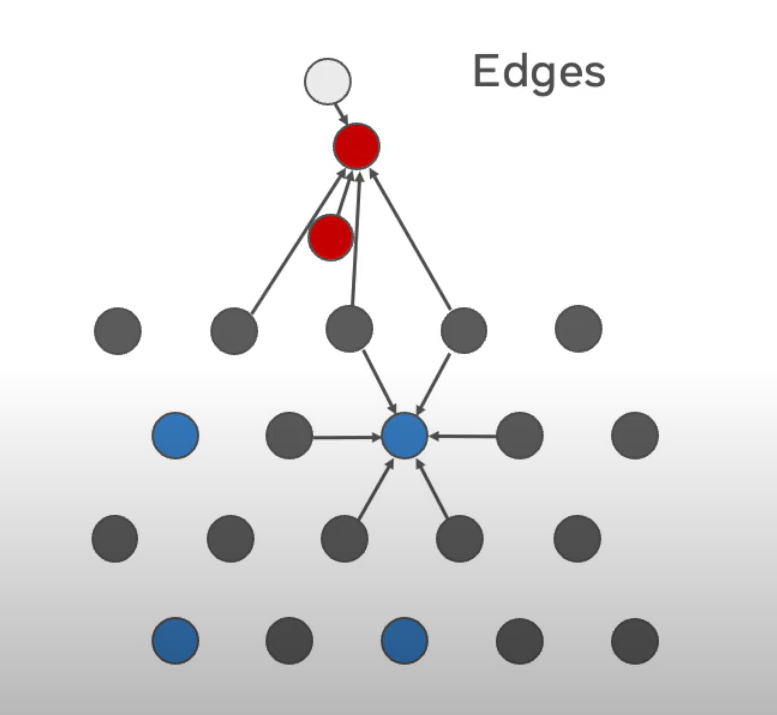
Fig 3: Visual Representation of GNN
D. Training
The model is trained on large datasets of atomic structures and their corresponding relaxed energies. The goal during training is to minimize the difference between the predicted relaxed energy and the true relaxed energy (as obtained from quantum mechanical simulations or other computational methods).
In IS2RE, this GNN-based approach allows the model to make efficient predictions about the stability of a material (in the form of relaxed energy) based on its initial structure, without needing to perform costly simulations.
VI. PROPOSED METHODOLOGY
In this project, we propose to develop a user-centric application that leverages machine learning (ML) models for material stability prediction and screening, specifically focusing on the relaxed energy prediction of materials in the context of catalyst design. The methodology involves integrating pre-trained models (IS2RE) into various functional modules of the application, allowing users to input material structures, screen potential candidates, and compare results for decision-making. Below is an outline of the methodology, detailing how ML will be applied across key modules.
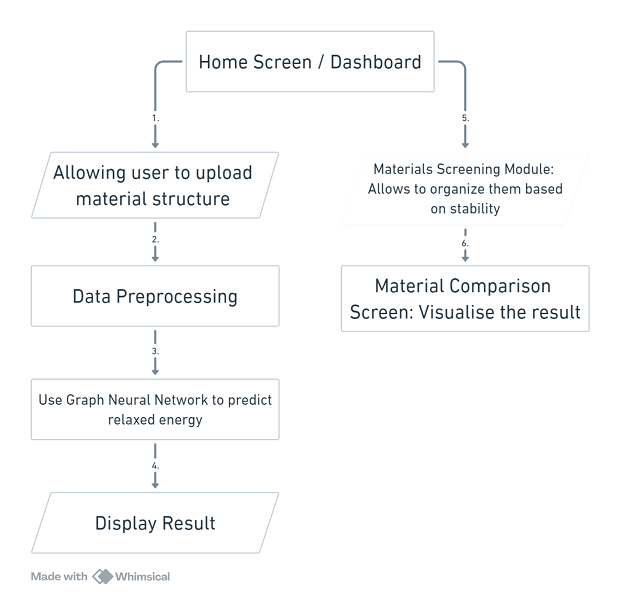
FIG 4: Application Flow Chart
A. Home Screen / Dashboard
- The Home Screen or Dashboard will serve as the entry point for users, providing access to material prediction tools, recent activity, and personalized recommendations. While no direct machine learning model is required for this module, we will implement a simple recommendation system to suggest materials based on user interaction history. This will enhance user experience by directing attention to materials that are of similar types or have shown promising stability in prior predictions.
- Data-drive Recommendations: Using a lightweight machine learning model, the system will analyse past predictions and suggest relevant material structures for further exploration or analysis.
B. Material Input Screen
In this module, users will be able to upload material structures in standardized formats such as CIF or XYZ files. These structures will be pre-processed to generate appropriate features for input into the machine learning model.
- Data Preprocessing: The uploaded atomic structures will be parsed using the Atomic Simulation Environment (ASE) library to extract relevant features such as atomic positions, types, and bond structures. These will be converted into a graph representation or tensor-based format suitable for the IS2RE model.
- File Upload & Processing Pipeline: A backend system (Flask/Django) will manage the file uploads, and preprocessing will take place on the server, where data is converted into the format required for model inference.
C. Results Screen (Relaxed Energy Prediction)
The core machine learning component of the application will reside in the Results Screen, where the predicted relaxed energy for a given material structure will be displayed.
- IS2RE Model: The IS2RE model will be utilized to predict the relaxed energy of the material based on its initial atomic structure. The model takes the pre-processed features (atomic positions, types) and directly predicts the relaxed energy, Erelaxed which is a key indicator of material stability.
- Model Inference & Display: The predicted energy values will be presented to the user along with relevant stability insights, such as thresholds for stable or unstable configurations. The prediction process will be streamlined for real-time feedback using pre-trained models hosted on the backend.
D. Materials Screening Module
The Materials Screening Module is designed to allow users to perform high-throughput screening of multiple materials, predicting their relaxed energies and ranking them based on stability.
- Batch Processing of Materials: Users can upload multiple material files at once (e.g., as a compressed ZIP file), and each material structure will undergo the same preprocessing and inference pipeline using the IS2RE model.
- Parallel Inference Pipeline: To ensure scalability and speed, the model inference for multiple structures will be performed in parallel, using multi-threaded or distributed computing techniques.
- Ranking Based on Stability: After predicting the relaxed energies for all uploaded materials, a ranking system will be implemented to order the materials from most to least stable. Materials with lower relaxed energies will be considered more stable and thus more suitable for catalyst applications.
E. Material Comparison Screen
The Material Comparison Screen will enable users to compare the predicted results of multiple materials side by side, focusing on the relative stability of different structures.
- Comparative Analysis: For materials selected by the user, their relaxed energy predictions will be displayed in a tabular or graphical format. Users will be able to compare the relaxed energies and other relevant properties such as atomic arrangements.
- Visualization Tools: Advanced visualization libraries (e.g., Plotly or D3.js) will be employed to generate comparative graphs or charts, allowing users to quickly identify trends and make informed decisions about material selection.
- ML-Driven Insights: In addition to visual comparisons, the system will provide machine learning-based insights, highlighting potential anomalies, patterns, or optimal choices based on the data at hand.
VII. FUTURE SCOPE
The proposed methodology for predicting relaxed energy in materials using the IS2RE dataset demonstrates significant potential for advancing materials science and engineering. Future work can focus on the following directions:
- Model Refinement and Enhancement: The current implementation of the IS2RE model serves as a foundational framework for predicting relaxed energies. Future research can explore the integration of advanced machine learning techniques, such as ensemble methods or transfer learning, to further improve prediction accuracy and generalization capabilities. Additionally, investigating the impact of incorporating additional features, such as electronic properties and crystal symmetry, may enhance the model's performance.
- Expansion of the Dataset: The accuracy of machine learning models is highly dependent on the diversity and volume of training data. Future studies could focus on expanding the IS2RE dataset by incorporating a wider variety of materials, particularly those that are currently underrepresented. Furthermore, generating synthetic data through simulations or augmenting existing data could facilitate improved model training and validation.
- Application to Real-World Problems: The developed framework can be applied to address practical challenges in materials design and discovery. Future research can investigate the use of the IS2RE model in high-throughput screening of novel materials for specific applications, such as catalysis, energy storage, or electronic devices. Implementing this model in collaboration with experimental teams can bridge the gap between computational predictions and real-world material performance.
- Integration with Other Predictive Models: Future endeavours could explore the integration of the IS2RE model with other predictive frameworks, such as those targeting structural stability or electronic properties. Developing a comprehensive predictive toolkit that encompasses multiple aspects of materials performance would provide a more holistic understanding of material behaviour.
- User Interface and Accessibility: Enhancing the user interface of the application to facilitate user interaction and accessibility is crucial. Future work could focus on developing a web-based platform that allows researchers and engineers to easily input material structures, visualize results, and obtain insights into material stability. This will democratize access to advanced predictive tools and encourage broader usage in both academic and industrial settings.
- Broader Applicability to Thermodynamic Properties: The models developed from the OC22 dataset could be extended to predict a broader range of thermodynamic properties beyond relaxed energies, including adsorption energies, reaction energies, and surface stability. Future research should focus on leveraging these predictions to facilitate the development of phase diagrams and other important thermodynamic analyses.
- Experimental Validation and Collaboration: Strengthening the collaboration between computational and experimental researchers will be crucial for validating computational predictions. Future efforts should focus on integrating computational findings with experimental data to enhance the reliability of models and ensure they accurately represent real-world scenarios.
Conclusion
In this project, we utilized advanced machine learning techniques, particularly graph neural networks (GNNs), to analyse the IS2RE dataset for predicting relaxed energies and understanding the behaviours of oxide materials. Our methodology demonstrated the effectiveness of GNNs in capturing the complexities of materials with long-range interactions, magnetic configurations, and charge balancing effects. The findings highlight the potential of these models to advance materials discovery in catalysis by accurately predicting thermodynamic properties and facilitating high-throughput screening of novel catalysts. By incorporating diverse datasets and innovative training strategies, we aim to improve model performance and applicability across various material systems. Additionally, we emphasize future research directions focused on enhancing data quality, model capabilities, and collaboration between computational and experimental domains, ultimately contributing to sustainable material development and bridging theoretical predictions with experimental validations. This work aspires to facilitate the discovery of new oxide catalysts and deepen our understanding of complex reaction mechanisms in materials science and engineering.
References
[1] Richard Tran, Janice Lan, Muhammed Shuaibi, Brandon M., Wood, Siddharth Goyal, Abhishek Das, Javier Heras-Domingo, Adeesh Kolluru, Ammar Rizvi, Nima Shoghi, Anuroop Sriram, F´elix Therrien, Jehad Abed, Oleksandr Voznyy,k Edward H. Sargent, Zachary Ulissi, and C. Lawrence Zitnick - “The Open Catalyst 2022 (OC22) Dataset and Challenges for Oxide Electrocatalysts” [2] Lowik Chanussot, Abhishek Das, Siddharth Goyal, Thibaut Lavril, Muhammed Shuaibi, Morgane Riviere, Kevin Tran, Javier Heras-Domingo, Caleb Ho, Weihua Hu, Aini Palizhati, Anuroop Sriram, Brandon Wood,k Junwoong Yoon, Devi Parikh, C. Lawrence Zitnick, and Zachary Ulissi –“The Open Catalyst 2020 (OC20) Dataset and Community Challenges” [3] Newell, R. G.; Raimi, D.; Villanueva, S.; Prest, B. Global Energy Outlook 2020: “Energy Transition or Energy Addition? With Commentary on Implications of the COVID-19 Pandemic; 2020”. [4] Kauwe, S. K.; Graser, J.; Vazquez, A.; Sparks, T. D. – “Machine learning prediction of heat capacity for solid inorganics. Integrating Materials and Manufacturing Innovation 2018”. [5] Schmidt, J.; Marques, M. R.; Botti, S.; Marques, M. A. – “Recent advances and applications of machine learning in solidstate materials science. npj Computational Materials 2019”. [6] Chandrasekaran, A.; Kamal, D.; Batra, R.; Kim, C.; Chen, L.; Ramprasad, R. “Solving the electronic structure problem with machine learning. npj Computational Materials 2019”. [7] Jamesh, M. I.; Sun, X. – “Recent progress on earth abundant electrocatalysts for oxygen evolution reaction (OER) in alkaline medium to achieve efficient water splitting – A review. Journal of Power Sources 2018.” [8] Gasteiger, J.; Shuaibi, M.; Sriram, A.; G¨unnemann, S.; Ulissi, Z.; Zitnick, C. L.; Das, A. GemNet-OC: “Developing Graph Neural Networks for Large and Diverse Molecular Simulation Datasets. Transactions on Machine Learning Research (TMLR) 2022” [9] Winther, K. T.; Hoffmann, M. J.; Boes, J. R.; Mamun, O.; Bajdich, M.; Bligaard, T. “Catalysis-Hub.org, an open electronic structure database for surface reactions. Scientific Data 2019.” [10] Godwin, J.; Schaarschmidt, M.; Gaunt, A. L.; Sanchez-Gonzalez, A.; Rubanova, Y.; Veli?ckovi´c, P.; Kirkpatrick, J.; Battaglia, P. “Simple gnn regularisation for 3d molecular property prediction and beyond. International Conference on Learning Representations. 2021.”
Copyright
Copyright © 2024 Vinod Ingale, Saniya Devale, Anushka Khedekar, Manasi Gaikwad, Dnyaneshwari More. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET65221
Publish Date : 2024-11-13
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online