

Ijraset Journal For Research in Applied Science and Engineering Technology
Karyotyping and Down Syndrome: Insights from Cytogenetic Analysis and Patient Data
Authors: Devika Mukherjee
DOI Link: https://doi.org/10.22214/ijraset.2024.62206
Certificate: View Certificate
Abstract
Down syndrome is one of the most commonly occurring chromosomal disorders, and in a rapidly growing population like India, cytogenetic evaluation is crucial for accurate diagnosis and medical intervention. Karyotyping remains the most relied upon cytogenetic analysis method due to its affordability and extensive experience. This paper provides an overview of Down syndrome, chromosomal aberrations, karyotype analysis, its requirements, and the processes involved. Additionally, it presents data collected in our laboratory over a year, facilitating data driven decision making. While Down syndrome has no cure, early detection through karyotyping enables better management and access to medical care, offering affected individuals the opportunity for a fulfilling life.
Introduction
I. INTRODUCTION TO DOWN’S SYNDROME
Down's syndrome is caused by trisomy of chromosome 21. The karyotype of a Downs syndrome patient shows an extra copy of chromosome 21. The type of Down’s syndrome determines the total number of chromosomes. Cytogenetic investigation should be performed on each patient suspected of having DS, since karyotyping is necessary for diagnostic confirmation, recurrence risk assessment, and genetic counseling. [1]
Individuals with Down’s Syndrome have distinct phenotypic appearance like:
- Flat back of the head
- Protruding tongue
- Extra skin present in the neck region
- Ears that are set low
- Gap between toes
- Umbilical hernia
- A solitary line running across the hand's palm (palmar crease)
- Epicanthal folds and flat face
- Smaller in stature as an adult and child
- Flat bridge of the nose
- Almond-shaped eyes with an upward slope
- A brief neck
- Tiny ears
- Little white patches on the eye's iris, or a portion of the eyes is differently coloured
- Tiny hands and feet relative to their body
- a solitary line running across the hand's palm (palmar crease)
- tiny pinky fingers that occasionally veer in the direction of the thumb
- Weak muscles and joints
- Significant gap between first and second toe
In addition to their distinct phenotypic appearance, individuals with Down syndrome are often predisposed to various health conditions. Children with Down syndrome frequently suffer from congenital heart diseases, necessitating regular medical intervention and frequent check-ups throughout their lives. Furthermore, they are genetically predisposed to leukemia and other blood cancers. Intestinal stenosis, characterized by the narrowing of the intestines, is also a common ailment among individuals with Down syndrome. Additionally, babies born with Down syndrome commonly experience umbilical hernias. [1,3]
In addition to their distinctive physical features, individuals with Down syndrome and their families face numerous challenges. Mental retardation is a common characteristic of individuals with Down syndrome, with the degree varying from mild to severe. The severity of the condition is typically assessed by a physiatrist through thorough analysis, and individuals may require psychological support, such as therapy, throughout their lives. [3]
Individuals with Down syndrome often need extra attention and care from family members, as well as at school. This support is essential not only for academic progress but also for physical fitness, as well as the development of communication and social skills. It is common for individuals with Down syndrome to achieve important milestones later than their peers. [3]
II. TYPES OF DOWN’S SYNDROME
Down’s syndrome is caused by trisomy of chromosome 21. It may be present in either of the 3 forms : Classical form, as a Robertsonian Translocation or in Mosaic form.A.
- Classical Down’s syndrome
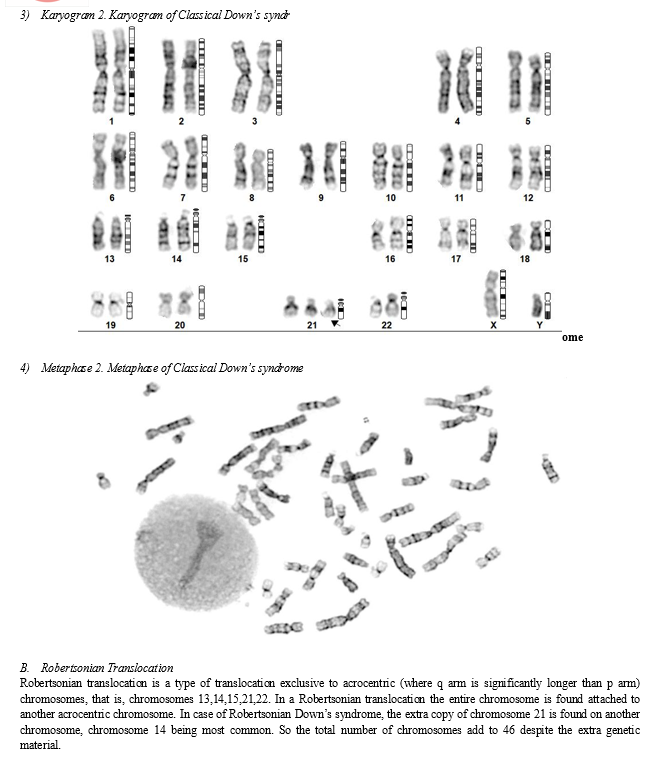
The karyotype of a person with classical down’s syndrome shows an extra copy of chromosome 21. Their total number of chromosomes add up to 47 instead of 46. [26,27,28]
Here the difference can be observed in the following karyogram.


C. Mosaic Down’s Syndrome
Mosaic Down syndrome occurs when there are two distinct cell populations or lineages in a female's ovary, each with a different chromosomal composition. In this condition, one of the cell populations contains an extra copy of chromosome 21, resulting in a total of 47 chromosomes, while the other cell population has the typical chromosomal makeup. [48,49]
Diagnosing mosaic Down syndrome is particularly challenging compared to other types of Down syndrome. This is primarily because the diagnostic process is more extensive and time-consuming for geneticists. To confirm a diagnosis of mosaic Down syndrome, geneticists must screen at least 30 metaphases, with a minimum of 5-6 metaphases showing trisomy 21. [48,49]
III. CAUSES OF DOWN'S SYNDROME
An individual with down's syndrome has either inherited it from a parent (even if they are phenotypically normal) or it has occurred without any previous history. The latter is known as ‘De Novo’ occurrence of Down’s syndrome. Most of the time their occurrence is De Novo.
A. Inherited Down’s Syndrome
Inherited Down’s Syndrome is also known as familial Downs syndrome. Only 5% of individuals with Down’s syndrome have inherited it from either one of their parents.
Usually one of the parents has a balanced translocation involving an additional copy or extra portion of chromosome number 21. Because there is an even exchange of the genetic material or a balanced translocation, the total number of chromosomes remain 46 despite the trisomy of chromosome 21. Another interesting thing to note is that individuals with balanced translocation are commonly phenotypically normal. However their children have a very high probab;ity of being born with Down’s syndrome. They also have high chance of having recurrent pregnancy losses or of having a bad obstetric history (RPL/OBD). [24,25,28]
B. De Novo Occurrence of Down’s syndrome
They are the most commonly occurring cause of Down’s syndrome. 95 percent of Individuals with Down's syndrome do not have familial history and their occurrence is indeed De novo. Hence they are also known as sporadic Downs syndrome. They occur due to errors during oogenesis- the process of egg formation or spermatogenesis-process of sperm formation. The mistakes made during gamete formation can be attributed to a variety of reasons. They can be classified as age of parents, especially advanced maternal age, environmental factors and nature of marriage such as consanguineous marriage. [24,25]
C. Inaccurate Oogenesis
Separation of chromosomes is an integral part of oogenesis as it ensures each ovum has 46 chromosomes of normal morphology.
To ensure that each egg has the appropriate number of chromosomes, the homologous chromosomes must separate correctly during meiosis 1 of oogenesis. Any error at this stage may lead to trisomy.
As women age, their likelihood of experiencing nondisjunction increases. Nondisjunction can be defined as a condition in which homologous chromosomes fail to split correctly during cell division. When nondisjunction occurs, eggs may carry an extra copy of chromosome 21. Trisomy of chromosome 21 causes Down syndrome in the progeny. [29,30]
Advanced maternal age, typically defined as women above the age of 35, is associated with an increased risk of having a child with Down syndrome. While the exact reasons for the higher incidence of chromosome separation errors in older women are not fully understood, researchers are studying changes in the structure and declining function of egg cells over time as potential contributing factors. It's essential to note, however, that age alone is not the sole determinant of the risk of having a child with Down syndrome. In fact, the majority of mothers who have children with Down syndrome are under the age of 35. This is because most women become mothers at a younger age, and thus, the majority of pregnancies occur in younger women. [29,30]
D. Problems in Spermiogenesis
In males, production of sperm or spermatogenesis continues throughout their lifetime. However as male age, the quality of sperm produced deteriorates.
Sperm motility reduces as men age. This affects their ability to fertilize ovum and produce healthy embryos with the correct number of chromosomes.
[31,32,33,34]
Sperm DNA can become damaged with advancing age as a result of oxidative stress, toxins in the environment, and unhealthy lifestyle choices like smoking, binge drinking and recreational drugs.
Chromosome abnormalities in sperm, such as aneuploidy, which is a common cause of Down syndrome can result from this DNA damage. The organization and structure of chromosomes in sperm cells may degrade with age. This raises the possibility of mistakes during cell division and the creation of sperm with abnormal numbers of chromosomes. Men become less capable of repairing DNA damage as they age, which additionally raises the possibility of chromosomal abnormalities.
[33,34,35,36]
Although sperm have a lower overall risk of causing chromosomal aberrations (including Down syndrome) than eggs do, there is a positive correlation between an increased risk of Down syndrome in offspring and advanced paternal age. Therefore, when determining the likelihood of chromosomal abnormalities in potential children, both the mother's and the father's age should be taken into account. [33,34]
E. Radiation and Mutation
Radiation and mutations are well known to alter DNA and disrupt cellular processes.
Exposure to them can disrupt the normal genetic processes involved in cell division, leading to chromosomal abnormalities such as trisomy of chromosome number 21. [47,48]
Ionizing radiation exposure, such as that from X-rays or gamma rays, can damage DNA by dissolving chemical bonds within the molecule. Cell division errors, such as nondisjunction, may arise from this damage if it happens during the development of female egg cells or male sperm cells. When chromosomes are unable to separate correctly during cell division, it is referred to as nondisjunction. This results in an incorrect chromosome distribution in the resulting cells. When chromosome 21 fails to fuse during the development of an egg or sperm, it can cause an embryo to develop with three copies of chromosome 21 rather than the normal two in the case of Down syndrome. [47,48]
Changes in the DNA sequence known as mutations can arise naturally or as a result of exposure to mutagenic substances (mutagenic substances can be defined as substances that cause mutation in nucleic acids), such as specific chemicals or environmental elements. Although trisomy 21 due to nondisjunction is a more frequent cause of Down syndrome than mutations affecting chromosome 21, they can still play a role in the condition's development. The distinctive traits of Down syndrome can result from mutations that cause an extra copy of chromosome 21 or changes in genes on chromosome 21 that interfere with normal development. [47,48]
In conclusion, mutations and radiation exposure can interfere with normal genetic processes related to chromosome segregation and cell division, which may result in the development of Down syndrome.
F. The Ambiguous Relationship of Down’s Syndrome with Ovarian Mosaicism
Ovarian mosaicism refers to the presence of two or more cell populations of different chromosomal makeup in the ovaries of a female. Given that it may impact the likelihood of having a child with Down syndrome, this condition is relevant.
The relationship between ovarian mosaicism and the risk of having a child with Down syndrome is still not fully understood and remains somewhat ambiguous. There is conflicting evidence regarding the risk of having a child with Down syndrome.Some researchers claim that women who have ovarian mosaicism might be more likely to produce ova that have chromosomal abnormalities, such as an extra copy of chromosome 21. This may make the risk of having a child with Down syndrome higher. Other research, however, has not discovered a strong correlation between ovarian mosaicism and the likelihood of developing Down syndrome. [29,30]
To fully comprehend the association between ovarian mosaicism and the likelihood of Down syndrome, more investigation is required. This relationship may become clearer through studies utilizing more sophisticated genetic techniques and larger populations. [29,30]
IV. ABOUT KARYOTYPE ANALYSIS
A. Cytogenetic Analysis and Conventional Karyotyping
Cytogenetics is the study of chromones to study abnormalities, mutations and determinants of phenotype. It is undertaken by a variety of examinations such as karyotype analysis and FISH. Cytogenetic examinations involve the inspection of chromosomes to detect abnormalities. Karyotype analysis is a popular method used for cytogenetic testing. It is a method doctor and researchers have relied on for many years. It can be used to detect various chromosomal aberrations including aneuploidy, polyploidy and structural abnormalities in chromosomes.
In a normal human cell, there are 23 pairs of chromosomes or a total 0f 46 chromosomes. Out of the 23 pairs one pair determines the biological sex of the individual and are hence known as the sex chromosomes. Normally they are XX for female phenotype and XY for male phenotype. The remaining 22 pairs of chromosomes are known as autosomes and are identical in both males and females. Aneuploidy occurs when the total number of chromosomes is not 46. There may be extra chromosomes (such as Down's syndrome, Klinefelter’s syndrome etc) or missing chromosomes (such as Turner’s syndrome, etc). [9]
Down’s syndrome is classified as an autosomal aneuploidy as it is caused by trisomy of chromosome 21. Another example of aneuploidy is Edward’s Syndrome caused by Trisomy 18. [9]
Structural abnormalities can also be identified by karyotype analysis. The higher the resolution, the easier it is to identify. However, the resolution of karyotype analysis is limited, making it challenging to detect smaller abnormalities. Karyotype analysis can typically detect abnormalities larger than 5-10 megabases.In cases where doubts persist after a normal karyotype analysis, other cytogenetic tests, such as Fluorescence In Situ Hybridization (FISH), are recommended to confirm or rule out abnormalities. Furthermore, in these kinds of situations, molecular tests like Chromosomal Microarray Analysis (CMA) are frequently conducted. CMA provides a more comprehensive assessment of the genome in comparison to karyotype analysis and can detect smaller chromosomal abnormalities.
Therefore, if there are concerns regarding the presence of structural abnormalities, further cytogenetic testing, such as FISH or CMA, may be necessary to obtain a conclusive diagnosis. However in most cases, it is not necessary.
(9)(10)
Structural abnormalities are of various types.
- Duplication: In duplication, a portion of a chromosome is repeated, resulting in the chromosome bearing extra bands. [12] All other chromosomes typically appear normal. For example, Duplication 15q syndrome involves an extra copy of the long arm (q) of chromosome 15. [13]
- Deletion: Deletion occurs when a portion of a chromosome is absent. The particular chromosome (or chromosomes) lacks some of its bands, while all other chromosomes appear normal. For example Cri-du-chat syndrome is caused by a deletion in the short arm of chromosome 5 (5p).[11,14,15]
- Translocation: Translocation is the transfer of genetic material from one chromosome to another or the exchange of genetic material between two distinct chromosomes. Translocation may be balanced or unbalanced.
This occurs when there is an even exchange of genetic material between chromosomes. For example, Acute Lymphoblastic Leukemia (ALL) is caused by a balanced translocation between chromosome 9 and chromosome 22, known as the Philadelphia chromosome.[17]
Unbalanced translocation takes place when a part of a chromosome gets attached to another chromosome, resulting in one chromosome having extra material while the other chromosome loses material. [16,17,18]
Translocations may also involve rearrangements within a single chromosome.For instance, a segment from the long arm may become attached to the short arm. [16]
- Inversion: Inversion occurs when a portion of the chromosome is inverted within the chromosome. This results in the chromosome appearing abnormal or unusual. [19]
- Insertion: Insertion involves the addition of genetic material from another chromosome. The source of the inserted material may vary and can sometimes be unknown. [20]
One thing to keep in mind is that these abnormalities are not exclusive but may be coupled together.
In Robertsonian Down Syndrome, a type of down’s syndrome, there is an additional chromosome and an unbalanced translocation. Extra copy of chromosome 21 is found on another acrocentric chromosome, commonly chromosome 14.
Cytogenetic testing is a reliable test that is performed in various situations, including:
- Solid organ malignancies
- Hematologic malignancies
- Congenital diseases
- Prenatal testing after abnormal biochemical screening or ultrasound findings
- Parents with multiple miscarriages (RPL or BOH) or significant findings in their pedigree analysis
- Postnatal testing for patients with mosaicism, intellectual disability, autism, or developmental delays
Conventional karyotype analysis has limitations. As discussed earlier it is unable to detect abnormalities between 5 to 10 megabases. Karyotype also requires a cell culture of live cells. Hence transport of blood samples in refrigerated state, storage of samples remain tedious processes that need to be undertaken. Many molecular tests and genomic sequencing methods do not require live cells. Moreover, culture of cells takes 72 hours. PCR and other sequencing methods do not require culture of cells and can produce results in 24 hours. Despite the limitations and availability of advanced technology, conventional karyotyping remains a widely popular choice in India. This is because it is a much more affordable option in comparison to New Generation Sequencing (NGS) and Chromosomal Microarray (CMA). Cost of Karyotype test is between 2500-5000 INR in India, whereas cost Of CMA is above 25000 INR.
Hence the majority of the population and doctors gravitate towards Karyotype Analysis.
Overall, cytogenetic testing plays a vital role in identifying chromosomal abnormalities and guiding clinical management in a variety of medical conditions in this country.
V. INTERNATIONAL SYSTEM FOR HUMAN CYTOGENOMIC NOMENCLATURE
The International System for Human Cytogenomic Nomenclature (ISCN) serves as a standardized framework for characterizing and documenting human chromosomal anomalies. It offers guidelines for naming and interpreting various types of chromosomal aberrations observed during cytogenetic analyses. Maintained by the International Standing Committee on Human Cytogenomic Nomenclature, ISCN ensures consistency and precision in reporting chromosomal irregularities, providing a universal language for cytogeneticists worldwide. This system encompasses numerical deviations, structural rearrangements, and complex chromosomal alterations, defining specific symbols and abbreviations for their representation. By adhering to ISCN guidelines, cytogeneticists can effectively communicate and interpret chromosomal abnormalities in clinical and research contexts, aiding in accurate diagnosis, genetic counseling, and scientific inquiry. [21,22,23]
VI. CULTURE AND HARVESTING OF CELLS FOR ANALYSIS (WET LAB PROCEDURES)
In cytogenetics samples of patient’s who are suspected to have Down’s Syndrome follows the standard procedure of a non leukemia disease for that particular sample type. The laboratory receives three types of samples from patients who opt for karyotype analysis: Peripheral blood (in Heparin), amniotic fluid, and Chorionic Villi Sample (CVS). Among these, Peripheral Blood is the most commonly received sample and is procured through non-invasive procedures.
A. Sample Handling
Upon arrival, each sample is labeled and recorded with an internal lab ID in addition to the sample ID. This is done both manually in a register as well as in the company’s computer database.
Accompanying each sample is a form containing relevant information, including the age and sex of the patient, reasons for referral by the doctor, complete medical history, doctor's prescription, phenotypic details, and suspected diagnosis.
This comprehensive information ensures that the laboratory has all necessary details to conduct accurate karyotype analysis and provide appropriate diagnosis and recommendations based on the results.
B. Culture of Cells
The Standard Operating Procedure (SOP) for processing a standard Peripheral Blood sample involves several precise steps to ensure accurate cell culture:
- Extraction of sample: 0.5 microliters of peripheral blood sample is extracted from the flask containing heparin.
- Transfer to Culture Flask: The extracted blood sample is transferred to a larger flask that provides ample space for cells to grow.
- Addition of Culture Media: 5 ml of culture media is added to the flask containing the blood sample.
- Incubation Period: The cell culture is allowed to incubate for a period of 72 hours.
This process ensures the optimal conditions for cell growth and proliferation, facilitating accurate cytogenetic analysis of the peripheral blood sample.
For each sample received, the laboratory undertakes a meticulous process to ensure accuracy and reliability. Two separate cultures, labeled A and B, are prepared as a precautionary measure. This dual culture approach significantly reduces the probability of process disruption and delays in the event of unexpected contamination.
Cell culture is considered crucial, as any error during this process could lead to contamination. It's essential to note that this step cannot be repeated in the case of a small quantity of sample, underscoring the importance of precision and care at every stage of the process.
C. Composition of Culture Media
- RPMI Media 1640
Roswell Park Memorial Institute, or RPMI, is the standard culture medium that is widely used in cytogenetics. RPMI, which is especially designed for the proliferation of mammalian cells, is a great choice for cell cultures that are leukemia- or non-leukemia-related. Because of its well-balanced composition, which creates the perfect environment for cell growth and maintenance, it is an essential part of cytogenetic laboratories all over the world.
2. Fetal Bovine Serum
An environment that is conducive to healthy and active cell division demands a consistent flow of resources and nutrients. The provision of vital nutrients to growing cells is the purpose of addition of fetal bovine serum (FBS). It guarantees the optimal growth and development of cells. Growth factors, hormones, and other vital elements that promote cell division and preserve cell viability in culture are abundant in fetal bovine serum (FBS). It is essential for maintaining the conditions required for proper cell development and activity in cell culture.
3. Antibiotic (penicillin)
The prevention of harmful bacterial contamination in culture media is prevented by the inclusion of antibiotics, such as penicillin. The integrity of the culture may be jeopardized by contamination, which could result in a failed culture leading to the inability to yield karyotype of patient. Antibiotics aid in maintaining the integrity and purity of the cell culture by preventing the growth of pathogenic bacteria, which makes cytogenetic research analysis and experimentation more successful.
4. PHA or Phytohemagglutinin
Peripheral blood cultures adhere to the standard operating protocol (SOP) of non-leukemia cultures. However, there is a distinct difference between leukemia and non-leukemia cultures. Non-leukemia cell cultures require a mitogen to induce metaphase and facilitate cell proliferation. Consequently, the culture for non-leukemia cells is referred to as a "Stimulated Culture".
In contrast, leukemia cell cultures, typically derived from bone marrow, do not require a mitogen. Therefore, they are referred to as "Unstimulated Cell Cultures". A mitogen, such as PHA (Phytohemagglutinin), is essential to "stimulate" growth or mitosis in cultures. Mitosis, induced by mitogens, leads to cell division, thereby increasing the cell count in the culture. This distinction in culture protocols ensures the optimal growth and analysis of both non-leukemia and leukemia cells in cytogenetic studies.
Cells are usually cultured for 72 hours in order to perform a karyotype analysis.
The incubator's temperature and other parameters are precisely controlled to resemble the physiological circumstances found in the human body. The temperature has been adjusted to roughly 37 degrees Celsius, which is the average human body temperature.
By keeping these parameters stable, cells can grow and multiply as best they can, creating an environment that is very similar to what is found inside the human body. The cell culture and ensuing karyotype analysis depend on this carefully regulated environment.
D. Harvesting of cells
After 72 hours sufficient growth has been achieved. The cells have to be harvested to reveal chromosome karyotype analysis. Ideally the harvesting room should mimic human body temperature and 40% humidity.
This is the standard procedure followed for harvesting of the cultured cells.
- Arresting Cells in Metaphase
Chromosomes are best observed in metaphase. This is primarily due to the formation of the equatorial plate or metaphase flare, where chromosomes align at the center of the cell, making them highly visible under the microscope. Chromosome abnormalities are most noticeable during metaphase, when the chromosomes are condensed and easily observable.
Colchicine or Colcemid is used for this purpose. It disrupts the normal process of cell division, leading to the accumulation of cells in metaphase, thus enhancing the visualization of chromosomes for accurate karyotype analysis.
Colchicine/colcemid is left to act on the cells for 45 minutes. Colchicine interferes with normal cell division during this period, causing a build-up of cells in metaphase.
Mechanism of Arresting cells in Metaphase
The mechanism of metaphase arrest by colchicine involves its interaction with microtubules, which are the support system for the normal process of cell division.
During cell division, microtubules form the mitotic spindle, which helps to segregate chromosomes into daughter cells. Colchicine disrupts this process by binding to tubulin, a protein that forms microtubules, and inhibiting microtubule polymerization. As a result, improper formation of the mitotic spindle impairs chromosome segregation. Colchicine treatment causes cells to become stuck in metaphase because it prevents them from continuing through mitosis. Colcemid binds to the tubules even faster than colchicine.
This metaphase arrest allows cytogeneticists to visualize and study cells at this particular stage of cell division making it easier to prepare and analyze chromosomes for karyotype analysis.
(5,6,7,8)
2. Centrifugation after Induction of Metaphase Arrest
After the addition of Colchicine or Colcemid the sample is left for 45 minutes for the colchicine/colcemid to act. The sample is then centrifuged for 10 mins at 1300 RPM (revolutions per minute).
In the centrifuge, the sample experiences two types of forces simultaneously: centripetal force and centrifugal force. The temperature inside the centrifuge is set to mimic human body conditions. It is set to 37 degree celsius.
3. Supernatant is discarded
4. Salting out of chromosomes
Hypotonic solution is added to serve two purposes. Firstly, it serves to "salt out" the chromosomes, separating them from other cellular components. Second, the hypotonic solution makes use of the osmosis principle. Chromosomes absorb water when they are in a hypotonic solution, which causes them to swell and become more visible under a microscope. This makes karyotype analysis easier.
A typical hypotonic solution is a potassium chloride (KCl) solution.This solution is prepared by precisely adding 0.56 grams of KCl salt to 100 ml of autoclaved or sterile distilled water.
To guarantee the hypotonic solution's effectiveness and to keep the ideal conditions for a precise karyotype analysis, it is prepared fresh every day. This ensures the efficacy of the solution.
Typically, 10 ml of KCl solution is added, and the cells are then incubated in the solution for 30 minutes.
5. Centrifugation
The cell suspension is centrifuged for 25–30 minutes at 37 degrees Celsius after the incubation period. Centrifugation aids in the separation of the enlarged cells from the supernatant, enabling the collection of cells for further processing and analysis in karyotype analysis.
6. Carney’s Fixation
Carney’s fixation is a commonly used fixative solution in cytogenetics. It consists of Methanol and Acetic acid in a 3:1 ratio.
- Methanol: Preserves cellular morphology.
- Acetic acid: Acts to lyse red blood cells (RBCs) by breaking down hemoglobin into dark brown hematite.
The cell pellet is mixed with 10 ml of Carney's fixative solution, and the mixture is centrifuged. The supernatant, containing cellular debris and unlysed RBCs, is then discarded.
This step is repeated until the solution runs clear, which is usually thrice. This indicates there is more RBCs and cell debris left for lysis.
7. Slide Preparation
After fixation is complete, slides are prepared for further analysis. Important information is written on the writable surface at the lower end of each slide. This includes the patient’s name, internal lab ID, and designation of whether the culture is A or B. As mentioned previously, two cultures are typically set up for each patient to ensure comprehensive analysis. Two slides are prepared for each culture to accommodate the duplicate cultures.
Additionally, a small portion of the sample is retained in the flask and refrigerated. This precautionary measure is taken in case an insufficient number of metaphases are observed during initial analysis, necessitating the preparation of new slides. However, it's important to note that this step may not be feasible if the sample quantity is inadequate. Even in the case of unanticipated events, this reliable method guarantees that all pertinent data is adequately recorded and that enough samples are available for thorough karyotype analysis. The supernatant of the flask is mostly discarded leaving behind the pellet. Using a dropper, the pellet is mixed with a small amount of the supernatant left behind and is carefully dropped onto a clean labeled slide. In order to guarantee appropriate metaphase dispersal and avoid chromosomal overlap, the pellet is dropped onto the slide at a considerable height, usually approximately 6 inches.
The purpose of dropping it from a height is to spread the metaphases to prevent overlap of chromosomes. This is a very important step in regard to observing it under the microscope. The slide is held at a 90 angle to prevent spillage. The slide is then kept on a hot plate which has a temperature set around 70 degrees.
In order to ensure that the chromosomes are released and properly spread out and heat fixed onto the slide for further analysis by karyotyping. The hot plate helps to evenly distribute the cell suspension on the slide and facilitates the drying process. This step is crucial to aid the microscope visualization of chromosomes to obtain high resolution results.
8. Aging of the Slides
After preparation, the slides undergo an aging process to ensure optimal fixation of the cells and chromosomes.
The slides can be aged using one of two methods:
- Overnight Aging
The slides are left overnight in a hot air oven set at 65 degrees Celsius. This gradual aging process allows for thorough drying and fixation of the cells and chromosomes.
- Fast Aging
The slides can be aged for an hour at a higher temperature of ninety degrees Celsius if there is a limited time to produce results. This accelerated aging process ensures rapid drying and fixation of the cells and chromosomes, allowing for quicker analysis if needed.
Both aging methods serve to ensure that the cells and chromosomes are properly fixed onto the slides, providing optimal conditions for accurate karyotype analysis.
9. Staining or Casting of slides
The GTC (Giemsa-Trypsin-Color) banding method, a commonly used technique in cytogenetics, is used to stain chromosomes. Giemsa stain and trypsin are combined in this method to provide the best possible chromosome visualization and banding.
Trypsin, an enzyme used for banding, plays a crucial role in this staining method. Trypsin acts on proteins and partially digests the AT-rich (Adenosine and Thymine) regions of the chromosomes. This digestion process results in the creation of distinct bands on the chromosomes, allowing for detailed analysis and interpretation. The GTC banding method, combining Giemsa stain and trypsin, enables cytogeneticists to obtain clear and highly informative banding patterns on chromosomes, facilitating accurate karyotype analysis and identification of chromosomal abnormalities.
E. Procedure
PBS buffer (Phosphate Buffered Saline) and 25 mg of trypsin are combined to create the trypsin solution. PBS buffer typically has a pH of 7.4, making it slightly alkaline. This solution is made from scratch everyday to guarantee its efficacy.
The aged slides are then immersed in the trypsin-PBS solution. Generally, the slides are dipped twice, but this decision may vary depending on the quality of the slides. The researcher must carefully observe and determine the number of dips necessary during this process. Trypsin treatment is followed by dipping the slide into chilled distilled water. This step serves to dissolve the trypsin solution in order to prevent the trypsin solution from completely fragmenting down the chromosomes. The cold temperature also helps minimize damage to the chromosomes. The slide is then submerged in Giemsa stain for approximately 2-3 minutes, although the duration may vary depending on the specific conditions. The scientist must use their judgment to determine the optimal staining time.
After staining, the slides are set on a slide tray and allowed to come to room temperature through air drying.
This GTG banding and drying process ensures that the chromosomes are properly stained and fixed onto the slides, allowing for accurate karyotype analysis.
F. Karyotype Analysis
The prepared slides can be observed under a fluorescence microscope following the wet lab processing. In this laboratory a semi-automated system manufactured by OLYMPUS is utilized for this purpose.
The slides are coated with immersion oil that has a refractive index of 1.5 before being observed. The chromosomes are then examined under 100X magnification, allowing for detailed visualization and analysis.
The microscope is connected to a computer system equipped with specialized software that is used for karyotype analysis. This software facilitates the visualization and analysis of chromosomes, allowing for the accurate identification of metaphase clusters and the creation of karyotype reports. This semi-automated system scans the prepared slides for detection of possible metaphase clusters using the microscope. Once selected, the microscope captures the chromosomes in each metaphase at 100X magnification. These captured metaphases are then added to the computer's database, where cytogeneticists can karyotype them to detect anomalies.
The resolution and clarity of the chromosomal bands are largely determined by the wet lab procedures, particularly slide preparation and banding. Any inconsistencies during these procedures can significantly impact the resolution and accuracy of karyogram.
Therefore, attention to detail and adherence to standard protocols during these wet lab procedures is imperative to a correct and reliable diagnosis. By upholding high standards in slide preparation and banding, cytogeneticists can achieve optimal resolution and clarity of chromosome bands, facilitating accurate diagnosis and karyotyping for patients. It emphasizes the importance of accuracy and diligence of the wet lab processes involved in cytogenetic analysis.
VII. DATA: CASE REPORTS
Here is a list of Down's syndrome cases from the month of September 2023 to 26 April 2024 diagnosed by this laboratory. These are cases from different parts of India. To respect confidentiality of the patients, their name and personal information have not been included.
|
Serial No. |
Age |
Gender |
Karyotype |
Type of Down Syndrome |
|
1 |
1 day |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
2 |
1 day |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
3 |
28 months |
Male |
47,XX,+21 |
Classical Down's Syndrome |
|
4 |
2 days |
Male |
47,XX,+21 |
Classical Down's Syndrome |
|
5 |
28 months |
Male |
47,XX,+21 |
Classical Down's Syndrome |
|
6 |
1 month |
Male |
47,XX,+21 |
Classical Down's Syndrome |
|
7 |
2 days |
Male |
47,XX,+21 |
Classical Down's Syndrome |
|
8 |
1 months |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
9 |
7 days |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
10 |
7 days |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
11 |
4 months |
Male |
47,XX,+21 |
Classical Down's Syndrome |
|
12 |
6 months |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
13 |
11 months |
Male |
47,XX,+21 |
Classical Down's Syndrome |
|
14 |
3 months |
Male |
47,XX,+21 |
Classical Down's Syndrome |
|
15 |
5 months |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
16 |
8 years |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
17 |
6 years |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
18 |
8 months |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
19 |
3 days |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
20 |
13 months |
Male |
47,XX,+21 |
Classical Down's Syndrome |
|
21 |
2 months |
Male |
47,XX,+21 |
Classical Down's Syndrome |
|
22 |
2 months |
Male |
47,XX,+21 |
Classical Down's Syndrome |
|
23 |
4 months |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
24 |
4 months |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
25 |
8 months |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
26 |
7 days |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
27 |
11 years |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
28 |
3 months |
Male |
47,XY,der(14;21)(q10;q10)+21 |
Robertsonian Translocation Down's Syndrome |
|
29 |
26 days |
Male |
47,XY,der(14;21)(q10;q10)+21 |
Robertsonian Translocation Down's Syndrome |
|
30 |
29 days |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
31 |
7 days |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
32 |
1 month |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
33 |
2 years |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
34 |
1 month |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
35 |
2 days |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
36 |
2 days |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
37 |
1 year |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
38 |
9 months |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
39 |
13 days |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
40 |
10 days |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
41 |
1 year |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
42 |
1 year |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
43 |
7 months |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
44 |
8 months |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
45 |
8 months |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
46 |
7 months |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
47 |
2 years |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
48 |
4 months |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
49 |
17 years |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
50 |
2 days |
Male |
46,XY,der(14;21)(q10;q10),+21 |
Robertsonian Translocation Down's Syndrome |
|
51 |
2 years |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
52 |
10 months |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
53 |
1 month |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
54 |
1 year |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
55 |
1 year |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
56 |
2 years |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
57 |
1 year |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
58 |
2 months |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
59 |
3 months |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
60 |
8 days |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
61 |
3 years |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
62 |
2 days |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
63 |
9 days |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
64 |
1 year |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
65 |
31 years |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
66 |
6 years |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
67 |
8 months |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
68 |
6 months |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
69 |
12 Days |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
70 |
2 Months |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
71 |
2 years |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
72 |
11 |
Male |
47,XX,+21 |
Classical Down's Syndrome |
|
73 |
1 Year |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
74 |
1w 3d |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
75 |
1 Year |
Female |
47,XY,+21 |
Classical Down's Syndrome |
|
76 |
3 months |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
77 |
6 months |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
78 |
1 Year |
Female |
47,XY,+21 |
Classical Down's Syndrome |
|
79 |
5 days |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
80 |
10 months |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
81 |
1 month |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
82 |
1 year |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
83 |
1 year |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
84 |
6 years |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
85 |
8 months |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
86 |
1 Year |
Female |
47,XX,+21 |
Classical Down's Syndrome |
|
87 |
5 months |
Male |
47,XY,+21 |
Classical Down's Syndrome |
|
88 |
23 weeks |
Male |
47,XY,+21 |
Classical Down's Syndrome |
VIII. DATA ANALYSIS
We can use this data to determine a variety of trends across. It is important to note that this data is small in size, consisting of only 88 patients and has been collected over a short period of time, that is, a year. Hence the limited time and number of cases makes it difficult to compare it to long term studies that have been extensively collected over a long period of time and have information about demographics. The particular demographics of people included in the data can't be listed, but it is from different regions of india.
However this data is still widely useful from the perspective of laboratories and to observe trends in smaller size data.
A. Patient Age
All ages have been converted to days to ensure uniformity :
1. 1 day = 1 day
2. 28 months = 840 days
3. 2 days = 2 days
4. 1 month = 30 days
5. 1 month = 30 days
6. 2 days = 2 days
7. 1 month = 30 days
8. 7 days = 7 days
9. 7 days = 7 days
10. 4 months = 120 days
11. 6 months = 180 days
12. 11 months = 330 days
13. 3 months = 90 days
14. 5 months = 150 days
15. 8 years = 2920 days
16. 6 years = 2190 days
17. 8 months = 240 days
18. 3 days = 3 days
19. 13 months = 390 days
20. 10 days = 10 days
21. 1 year = 365 days
22. 1 year = 365 days
23. 7 months = 210 days
24. 8 months = 240 days
25. 8 months = 240 days
26. 7 months = 210 days
27. 2 years = 730 days
28. 4 months = 120 days
29. 17 years = 6205 days
30. 2 days = 2 days
31. 2 years = 730 days
32. 10 months = 300 days
33. 1 month = 30 days
34. 1 year = 365 days
35. 1 year = 365 days
36. 2 years = 730 days
37. 1 year = 365 days
38. 2 months = 60 days
39. 3 months = 90 days
40. 8 days = 8 days
41. 3 years = 365 days
42. 2 days = 2 days
43. 9 days = 9 days
44. 1 year = 365 days
45. 31 years = 11315 days
46. 6 years = 2190 days
47. 8 months = 240 days
48. 6 months = 180 days
49. 12 days = 12 days
50. 2 months = 60 days
51. 2 years = 730 days
52. 11 months = 330 days
53. 1 year = 365 days
54. 1 year = 365 days
55. 1 week 3 days = 10 days
56. 1 year = 65 days
57. 3 months = 90 days
58. 6 months = 180 days
59. 1 year = 365 days
60. 5 days = 5 days
61. 10 months = 300 days
62. 1 month = 30 days
63. 1 year = 365 days
64. 1 year = 365 days
65. 6 years = 2190 days
66. 8 months = 240 days
67. 1 year = 365 days
68. 5 months = 150 days
69. 23 weeks fetus = 161 days
70. 4 days = 4 days
71. 4 months = 120 days
72. 2 months = 60 days
73. 3 years = 1095 days
74. 2 days = 2 days
75. 9 days = 9 days
76. 8 days = 8 days
77. 6 months = 180 days
78. 12 days = 12 days
79. 2 months = 60 days
80. 2 years = 730 days
81. 11 months = 330 days
82. 1 year = 365 days
83. 1 year = 365 days
84. 6 years = 2190 days
85. 1 year = 365 days
86. 1 year = 365 days
87. 5 months = 150 days
88. 6 months = 180 days
Calculation of the mean, median, and standard deviation.
B. Average Age of Patient when Diagnosed with Down’s Syndrome
Mean age = sum of Ages/Number of Ages
= 32,060/88
= 364.3181≈ 365 days
The average age at which a patient is diagnosed is 365 days or one year.
???????C. Median Age of Patient when diagnosed with Down's Syndrome
Since there are 88 ages or data points listed, the median will be the average of the 44th and 45th values when the data is arranged in ascending order.
Since the data is sorted, the 44th and 45th values are both 365.
So, the median is 365 days.
D. Standard Deviation
A measure of the degree of variation or dispersion in a set of values is the standard deviation (SD). Within the framework of patient age in this instance, the standard deviation indicates the degree of variance or dispersion in the patient ages.
A low standard deviation, on the other hand, suggests that the ages are closer to the mean, whereas a high standard deviation suggests that the ages are more dispersed from the mean.
To conclude, the standard deviation indicates the degree to which a given value deviates from the average (mean) of all the values.

So, the mean age is 365 days, the median age is 365 days, and the standard deviation is approximately 54.92 days.
E. Sex Disparity
Down syndrome affects people of all ages and races, with very little variation in prevalence between males and females. As a result, neither sex is thought to be more vulnerable to Down syndrome or to receive a diagnosis of it more frequently. Although some argue that there is a small gender difference in the prevalence of Down syndrome. Some research has shown a slightly higher prevalence in men even though both men and women can be impacted by the illness.
[37,38,39]
In this data consisting of total of 88 patients,
55 out of 88 patients are MALES.
33 out of 88 are FEMALE.
Hence, the percentage of males is 62.50% and the percentage of females is 37.50%. In this data, it is more typical in MALES.
F. Type of Down’s Syndrome
85 out of 88 are cases of Classical Down's Syndrome
3 out of 88 are cases of Robertsonian Translocation
No cases of Down’s syndrome in Mosaic form have been reported in this laboratory till date.
Percentage of Classical Down's Syndrome cases = (85 / 88) * 100 ≈ 96.59%
Percentage of Robertsonian Translocation cases = (3 / 88) * 100 ≈ 3.41%
Most extensive long term studies have concluded that around 95% of individuals have Classical Down’s Syndrome karyotype. Robertsonian Translocation is the karyotype of 3%-4% of all cases. Less than 1% of cases appear in mosaic form. [1,39,40]
In this case, approximately 96.59% of Down’s Syndrome cases are cases of Classical Down’s Syndrome and 3.41% are caused by Robertsonian translocation.
???????IX. EARLY DETECTION TESTS
In order to identify Down syndrome at an early stage, early detection tests are necessary. Although there is no way to prevent Down syndrome, early detection enables parents to make informed decisions about their pregnancy, including seeking a medical termination if they choose to do so.
Caring for a child with Down's Syndrome is both financially expensive and mentally taxing for families. Thankfully, there are plenty of highly accurate and non-invasive early detection tests available for Down syndrome. These tests are widely promoted through government initiatives and awareness campaigns and are important components of prenatal care. To guarantee that expectant parents have access to these crucial tests, both public and private hospitals frequently incorporate these exams into their typical prenatal care packages.
A. The Double Marker and Triple Marker tests
The Double Marker and Triple Marker tests are two examples of these tests, which are usually conducted at particular phases of pregnancy. In order to determine the fetus's risk of Down syndrome and other chromosomal abnormalities, these tests examine blood samples from the mother. [41,42]
In the double marker test, only two hormones are tested at 10 to 13 week of pregnancy :
- Free beta-HCG
- PAAP – A
In the triple marker test, three hormones are tested at 14 to 20 week pregnancy:
- Alpha-fetoprotein (AFP)
- Unconjugated estriol
- beta-HCG [41,42]
The placenta's concentrations of three vital substances are measured:
- Alpha-Fetoprotein (AFP): This protein is made by the developing foetus. Elevations in this protein may indicate the presence of abnormalities such as neural tube defects, among others. [41,42]
- Chorionic gonadotropin (HCG) in humans: This hormone is produced by the placenta, and low levels of it may signal pregnancy hazards such as ectopic pregnancy or miscarriage. [41,42]
- Estriol is a type of estrogen produced by the placenta and the fetus. When low levels of AFP and high levels of HCG are paired, low estriol levels may indicate a higher chance of having a child with down syndrome. [41,42]
Abnormal levels of these substances can also indicate Down syndrome or Edward’s syndrome.
Accuracy and procedure of both the tests are very similar. The difference lies in time of testing and the markers being tested. There is an additional marker in the triple marker test. [41,42]
If these features are Observed in the fetus amniocentesis is recommended for further investigation.
B. Noninvasive Prenatal testing or NIPT
The noninvasive prenatal testing (NIPT) or noninvasive prenatal screening (NIPS) is a novel technique for determining a fetus's risk of specific genetic abnormalities. In contrast to conventional prenatal testing techniques, NIPT examines minute pieces of cell-free DNA (cfDNA) that are in circulation within the pregnant patient's bloodstream. When cells die and degrade, these cfDNA fragment, which usually have fewer than 200 DNA base pairs, are discharged into the bloodstream.
The main application of NIPT is the detection of chromosomal disorders brought on by an extra or missing copy (aneuploidy) of a chromosome. Due to its extra copy of chromosome 21, Down syndrome (trisomy 21) is one of the most frequently screened chromosomal abnormalities. [41,43,44]
- Accuracy
The Double Marker or Triple Marker tests are generally thought to be a lot more accurate than the NIPT. Despite the fact NIPT is still considered reliable in identifying specific chromosomal abnormalities, specifically Down syndrome. [41,43,44]
2. Major Limitations
- NIPT may not accurately reflect the baby's chromosomal makeup in cases of mosaic Down syndrome. Mosaic Down syndrome occurs when some fetal cells have the extra chromosome 21, while others do not. As NIPT analyzes cfDNA circulating in the maternal bloodstream, it may not detect the abnormality in cases where the affected cells are not well represented. [41,43,44]
- NIPT often cannot detect balanced translocation, a type of chromosomal abnormality where a reciprocal of genetic material between chromosomes occurs without an increase in total number of chromosomes. [41,43,44]
- NIPT may not always provide a conclusive answer, and further testing may be required to confirm the presence or absence of chromosomal abnormalities.
- It is unable to diagnose Mosaic Down’s syndrome. [41,43,44]
Despite these limitations, NIPT remains a valuable tool in prenatal care, offering expectant parents a noninvasive and relatively accurate method of screening for certain genetic abnormalities in the fetus.
???????C. Ultrasound
Ultrasound has significantly improved early diagnosis rates for Down syndrome. It is the most non-invasive early diagnosis method and a regular part of every pregnancy. Doctors look for certain characters that may indicate a Down’s syndrome fetus. This advancement has revolutionized prenatal screening, allowing for earlier detection and allowing informed decision making for expectant parents than ever before. [45,46]
Interestingly, ultrasound anomalies are more commonly observed in younger mothers, as compared to older (above 35 years of age). [45,46]
Common characteristics found in Ultrasound:
An elevated risk of Down syndrome is linked to several ultrasonography findings:
- Cystic hygroma (CH): An unusual sac filled with fluid that is visible behind the fetus's neck.
- Increased nuchal translucency (NT): A thickened area at the back of the fetus's neck, which can indicate an increased risk of chromosomal abnormalities.
- Abnormal ductus venosus (DV) flow: An irregular fetal vein's blood flow that may point to cardiac problems.
- Absent nasal bone: The absence of a nasal bone can be a warning sign of Down syndrome.
[45,46]
Major structural malformations and soft markers:
Apart from the aforementioned discoveries, additional ultrasonography indicators linked to Down syndrome consist of:
- Hyperechogenic bowel: The foetal bowel's increased brightness, which may be a sign of digestive problems.
- Choroid plexus cysts: Brain cavities filled with fluid.
- Ventriculomegaly: Enlargement of the ventricles in the developing brain.
- Cardiac defects: Defects in the foetal heart's structure.
[45,46]
These ultrasound results are crucial markers for additional diagnostic exams and guidance, enabling medical professionals to provide expectant parents with all encompassing care and assistance. [45,46]
X. MANAGEMENT OF DOWN’S SYNDROME
Specialized recommendations and healthcare guidelines have been established by the American Academy of Pediatrics (AAP) and the National Down Syndrome Society (NDSS) for parents, caregivers, and individuals with Down syndrome. These recommendations are essential for several reasons:
For parents or guardians caring for children with Down syndrome, these specific recommendations help define necessary care, enabling them to inform their primary care physicians about advised procedures and protocols.
For medical professionals, these guidelines outline the medical risks associated with Down syndrome and the necessary tests and procedures required for individuals with the condition. This ensures that healthcare providers are equipped with the knowledge and tools needed to provide optimal care for their patients with Down syndrome. [3]
As per May 5, 2022 the volume 149 of ‘Health Supervision for Children Down Syndrome’, the following examinations need to be undertaken timely [4]:
Table 1. Test/examination for Down’s Syndrome
|
S. No. |
Test/Exam |
Frequency |
|
1 |
TSH and T4-Thyroid Function Test |
Annual |
|
2 |
Auditory testing |
Every 2 years |
|
3 |
Cervical spine x-rays |
As needed for sports |
|
4 |
Ophthalmologic exam |
Every 2 years |
|
5 |
Echocardiogram-ECHO |
As indicated |
|
6 |
Mammography |
Baseline at 40 years, then every other year until 50, then annually |
|
7 |
Pap smear and pelvic exam |
Every 1-3 years after first intercourse |
|
8 |
Single finger bimanual exam |
If not sexually active, or unable to perform pelvic ultrasound |
|
9 |
Pelvic ultrasound |
Every 2-3 years |
|
10 |
Breast exam |
Annually |
|
11 |
General physical/neurological exam |
Routine adult care |
|
12 |
Clinical evaluation for sleep apnea |
As needed |
|
13 |
Diet and Exercise |
Low calorie, high-fiber diet. Regular exercise. Monitor for obesity. |
|
14 |
Health, abuse-prevention, and sexuality education |
Regularly |
|
15 |
Clinical evaluation of functional abilities |
Monitor for loss of independent living skills |
|
16 |
Neurological referral for early symptoms of dementia |
Decline in function, memory loss, ataxia, seizures, incontinence of urine and/or stool |
|
17 |
Monitor for behavior/emotional/mental health |
Regularly, with psych referral as needed |
|
18 |
Speech and language therapy |
As indicated |
In order to help doctors provide the best care possible for people with Down syndrome, the GLOBAL Medical Care Guidelines for people with Down Syndrome include evidence-based medical recommendations. In order to guarantee that they receive care that satisfies the best-practice standards for adults with Down syndrome, it is crucial that caregivers and individuals with Down syndrome go over these criteria with their clinicians. [3]
References
[1] Ahmed, I., Ghafoor, T., Samore, N. A., & Chattha, M. N. (2007). Down syndrome: Clinical and cytogenetic analysis. Journal of the College of Physicians and Surgeons Pakistan, 17(3), 159-162. [https://www.researchgate.net/profile/Tariq-Ghafoor/publication/7566992_Down_syndrome_Clinical_and_cytogenetic_analysis/links/56abab2908aeaa696f29dbb5/Down-syndrome-Clinical-and-cytogenetic-analysis.pdf](https://www.researchgate.net/profile/Tariq-Ghafoor/publication/7566992_Down_syndrome_Clinical_and_cytogenetic_analysis/links/56abab2908aeaa696f29dbb5/Down-syndrome-Clinical-and-cytogenetic-analysis.pdf) [2] Kava, M., Tullu, M. S., Muranjan, M., & Campeau, P. M. (2004). Down syndrome. Archives of Medical Research, 35(1), 66-69. [https://doi.org/10.1016/j.arcmed.2003.06.005](https://doi.org/10.1016/j.arcmed.2003.06.005) [3] National Down Syndrome Society (NDSS). Healthcare Guidelines. [https://ndss.org/resources/healthcare-guidelines#:~:text=Auditory%20testing%20(every%202%20years,out%20mitral%2Faortic%20valve%20problems](https://ndss.org/resources/healthcare-guidelines#:~:text=Auditory%20testing%20(every%202%20years,out%20mitral%2Faortic%20valve%20problems). [4] Broderick, K. E., Khan, J. R., & Davis, K. (2021). Health Supervision for Children and Adolescents. Pediatrics, 149(5), e2022057010. [https://publications.aap.org/pediatrics/article/149/5/e2022057010/186778/Health-Supervision-for-Children-and-Adolescents](https://publications.aap.org/pediatrics/article/149/5/e2022057010/186778/Health-Supervision-for-Children-and-Adolescents) [5] Gupta, S. C., Kim, J. H., Prasad, S., Patchva, S., & Aggarwal, B. B. (2013). Discovery of curcumin, a component of golden spice, and its miraculous biological activities. Clinical and Experimental Pharmacology & Physiology, 39(3), 283–299. [https://doi.org/10.1111/1440-1681.12014](https://doi.org/10.1111/1440-1681.12014) [6] Vale, R. D. (1987). The molecular motor toolbox for intracellular transport. Cell, 49(1), 1–4. [https://doi.org/10.1016/0092-8674(87)90747-1](https://doi.org/10.1016/0092-8674(87)90747-1 [7] Inoué, S. (1953). Molecular organization of mitotic spindle fibers. I. Demonstration of taxin, a 220,000 molecular weight protein unit in spindle fibers. The Journal of Cell Biology, 16(2), 227–237. [https://doi.org/10.1083/jcb.16.2.227](https://doi.org/10.1083/jcb.16.2.227) [8] Hauschka, T. S., & Villanueva, A. R. (1966). [Cytochemical studies of mammalian tissues. II. Isolation of nuclei and nucleoli from rat liver.] (https://pubmed.ncbi.nlm.nih.gov/5938902/#:~:text=Colcemid%20binds%20to%20tubulin%20much,for%20at%20least%201%20h.). Experimental Cell Research, 43(2), 359–367. [9] Matzuk, M. M., Burns, K. H., Viveiros, M. M., & Eppig, J. J. (2002). Intercellular communication in the mammalian ovary: oocytes carry the conversation. Science, 296(5576), 2178–2180. [https://europepmc.org/article/nbk/nbk563293](https://europepmc.org/article/nbk/nbk563293) [10] Wyandt, H. E., & Tonk, V. S. (2006). [ISCN 2005: an international system for human cytogenetic nomenclature](https://karger.com/books/book/358/ISCN-2020An-International-System-for-Human). Basel: S. Karger AG. [11] Shaffer, L. G., McGowan-Jordan, J., & Schmid, M. (Eds.). (2013). [ISCN 2013: An International System for Human Cytogenetic Nomenclature (2013th ed.)](https://www.karger.com/Book/Home/255248). Basel: S. Karger AG. [12] McGowan-Jordan, J., Simons, A., & Schmid, M. (Eds.). (2016). [ISCN 2016: An International System for Human Cytogenomic Nomenclature (2016th ed.)](https://www.karger.com/Book/Home/275452). Basel: S. Karger AG. [13] Sherman, S. L., & Allen, E. G. (2007). Examining the relationship between maternal age and the frequency of Down syndrome births: a new method. Cytogenetic and Genome Research, 117(1-4), 313-315. [https://doi.org/10.1159/000103196](https://doi.org/10.1159/000103196) [14] Sherman, S. L., Freeman, S. B., & Allen, E. G. (2005). Complex segregation analysis of trisomy 21 in Down syndrome families. Genetics in Medicine, 7(2), 131-138. [https://doi.org/10.1097/01.GIM.0000153664.79027.2A](https://doi.org/10.1097/01.GIM.0000153664.79027.2A) [15] Down, J. L. H. (1866). Observations on an ethnic classification of idiots. Clinical Lectures and Reports by the Medical and Surgical Staff of the London Hospital, 3, 259-262. [16] Pletcher, B. A., Toriello, H. V., & Noblin, S. J. (2020). Down Syndrome. In Adam MP, Ardinger HH, Pagon RA, et al. (Eds.), GeneReviews®. Seattle: University of Washington, Seattle. [https://www.ncbi.nlm.nih.gov/books/NBK1152/](https://www.ncbi.nlm.nih.gov/books/NBK1152/) [17] Allen, E. G., Freeman, S. B., Druschel, C., Hobbs, C. A., O\'Leary, L. A., Romitti, P. A.,... & Sherman, S. L. (2009). Maternal age and risk for trisomy 21 assessed by the origin of chromosome nondisjunction: a report from the Atlanta and National Down Syndrome Projects. Human Genetics, 125(1), 41-52. [https://doi.org/10.1007/s00439-008-0603-0](https://doi.org/10.1007/s00439-008-0603-0) [18] Velloso, E. P., Alvim, C. G., & Machado, C. M. (2014). Cri du chat syndrome. Jornal de Pediatria, 90(3), 259-263. [https://doi.org/10.1016/j.jped.2013.12.007](https://doi.org/10.1016/j.jped.2013.12.007) [19] Ledbetter, D. H., & Riccardi, V. M. (1982). Association of an 11;22 translocation with meningioma, melanoma, and “normal” offspring. Journal of Medical Genetics, 19(2), 126-127. [https://doi.org/10.1136/jmg.19.2.126](https://doi.org/10.1136/jmg.19.2.126) [20] Infantino, G. M., & Ricci, B. (2017). Cri-du-chat syndrome: Clinical profile and chromosomal microarray analysis in six patients. European Journal of Medical Genetics, 60(1), 39-44. [https://doi.org/10.1016/j.ejmg.2016.10.015](https://doi.org/10.1016/j.ejmg.2016.10.015) [21] Rowley, J. D. (1973). A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature, 243(5405), 290-293. [https://doi.org/10.1038/243290a0](https://doi.org/10.1038/243290a0) [22] Jee, Y. H., & Jung, J. S. (2018). Down syndrome: An overview. Endocrinology and Metabolism, 33(2), 123-129. [https://doi.org/10.3803/EnM.2018.33.2.123](https://doi.org/10.3803/EnM.2018.33.2.123) [23] Hassold, T., & Hunt, P. (2001). To err (meiotically) is human: the genesis of human aneuploidy. Nature Reviews Genetics, 2(4), 280-291. [https://doi.org/10.1038/35066065](https://doi.org/10.1038/35066065) [24] Hook, E. B., & Cross, P. K. (1983). Maternal age, gestational age, and rates of Down syndrome at livebirth: data from the New York State chromosome registry. American Journal of Human Genetics, 35(1), 190-196. [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1685477/](https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1685477/) [25] Thomas, N. S., Hassold, T. J., & Hunt, P. A. (2009). Molecular basis of maternal age-related trisomy in Down syndrome. Birth Defects Research Part A: Clinical and Molecular Teratology, 85(4), 326-327. [https://doi.org/10.1002/bdra.20584](https://doi.org/10.1002/bdra.20584) [26] Glaser, R. L., & Broman, K. W. (2006). Maternal age and risk for trisomy 21 assessed by the origin of chromosome nondisjunction: a report from the Atlanta and National Down Syndrome Projects. Human Genetics, 120(6), 869-878. [https://doi.org/10.1007/s00439-006-0274-8](https://doi.org/10.1007/s00439-006-0274-8) [27] Wyrobek, A. J., & Eskenazi, B. (2000). Semen quality in a residential andrology research cohort: age effect. Reproductive Toxicology, 14(4), 303-314. [https://doi.org/10.1016/S0890-6238(00)00102-6](https://doi.org/10.1016/S0890-6238(00)00102-6) [28] Singh, N. P., Muller, C. H., Berger, R. E., & Diehl, J. (2003). Effects of age on DNA double-strand breaks and apoptosis in human sperm. Fertility and Sterility, 80(6), 1420-1430. [https://doi.org/10.1016/S0015-0282(03)02127-6](https://doi.org/10.1016/S0015-0282(03)02127-6) [29] Eskenazi, B., & Wyrobek, A. J. (2003). Paternal age and birth defects: how strong is the association?. Occupational and Environmental Medicine, 60(8), 501-502. [https://doi.org/10.1136/oem.60.8.501](https://doi.org/10.1136/oem.60.8.501) [30] Robinson, D., & Kleinman, K. P. (2012). Understanding the effect of paternal age and birth order on the incidence of Down syndrome using data from the National Down Syndrome Project. Genet Epidemiol, 36(5), 452-460. [https://doi.org/10.1002/gepi.21643](https://doi.org/10.1002/gepi.21643 [31] Sherman, S. L., Allen, E. G., Bean, L. H., & Freeman, S. B. (2007). Epidemiology of Down syndrome. Mental retardation and developmental disabilities research reviews, 13(3), 221-227. [https://doi.org/10.1002/mrdd.20163](https://doi.org/10.1002/mrdd.20163) [32] Morris, J. K., & Springett, A. L. (2013). The National Down Syndrome Cytogenetic Register for England and Wales: 2010-2011 Annual Report. The National Down Syndrome Cytogenetic Register. [33] Vi?i?, A., Hafner, T., Vlatkovi?, I. B., Kora?, P., Habek, D., & Stipoljev, F. (2006). Prenatal diagnosis of Down syndrome: A 13-year retrospective study. Collegium antropologicum, 30(3), 555-558. [34] Natoli, J. L., Ackerman, D. L., McDermott, S., & Edwards, J. G. (2012). Prenatal diagnosis of Down syndrome: a systematic review of termination rates (1995–2011). Prenatal Diagnosis, 32(2), 142-153. [https://doi.org/10.1002/pd.2910](https://doi.org/10.1002/pd.2910) [35] Sifakis, S., Papantoniou, N., Kappou, D., & Antsaklis, A. (2015). Noninvasive prenatal diagnosis of Down syndrome: current knowledge and novel insights. Journal of Pregnancy, 2015, 1-9. [https://doi.org/10.1155/2015/624792](https://doi.org/10.1155/2015/624792) [36] Verma, L., Macdonald, F., Leedham, P., McConachie, M., & Hultén, M. (2000). Rapid and simple prenatal DNA diagnosis of Down\'s syndrome. Lancet, 356(9237), 1610-1611. [https://doi.org/10.1016/S0140-6736(05)72051-9](https://doi.org/10.1016/S0140-6736(05)72051-9) [37] Benn, P., & Cuckle, H. (2015). Theoretical performance of non-invasive prenatal testing for chromosome imbalances using counting of cell-free DNA fragments in maternal plasma. Prenatal Diagnosis, 35(3), 236-241. [https://doi.org/10.1002/pd.4460](https://doi.org/10.1002/pd.4460) [38] Norton, M. E., Jacobsson, B., Swamy, G. K., Laurent, L. C., Ranzini, A. C., Brar, H.,... & Tomlinson, M. W. (2015). Cell-free DNA analysis for noninvasive examination of trisomy. New England Journal of Medicine, 372(17), 1589-1597. [https://doi.org/10.1056/NEJMoa1407349](https://doi.org/10.1056/NEJMoa1407349) [39] Norton, M. E., Baer, R. J., Wapner, R. J., Kuppermann, M., Jelliffe-Pawlowski, L. L., Currier, R. J.,... & Ryckman, K. K. (2015). Cell-free DNA vs sequential screening for the detection of fetal chromosomal abnormalities: A randomized clinical trial. Jama, 314(21), 2241-2248. [https://doi.org/10.1001/jama.2015.13136](https://doi.org/10.1001/jama.2015.13136) [40] Odibo, A. O., Gray, D. L., Dicke, J. M., Stamilio, D. M., Macones, G. A., & Crane, J. P. (2008). Revisiting the fetal nasal bone: should we incorporate the fetal nasal bone into routine first?trimester sonographic examination? Ultrasound in Obstetrics and Gynecology: The Official Journal of the International Society of Ultrasound in Obstetrics and Gynecology, 32(2), 190-194. [https://doi.org/10.1002/uog.5307](https://doi.org/10.1002/uog.5307) [41] Antonarakis, S. E., Petersen, M. B., McInnis, M. G., Adelsberger, P. A., Schinzel, A. A., & Binkert, F. (1987). The meiotic stage of nondisjunction in trisomy 21: determination by using DNA polymorphisms. American Journal of Human Genetics, 41(2), 142-148. [https://doi.org/10.1016/S0002-9297(16)90438-5](https://doi.org/10.1016/S0002-9297(16)90438-5) [42] Madan, K., & Nieuwint, A. W. (2004). A comparison of the quality of prenatal diagnosis for trisomy 21 in different racial groups after the introduction of a national prenatal screening programme. BJOG: An International Journal of Obstetrics & Gynaecology, 111(12), 1444-1446. [https://doi.org/10.1111/j.1471-0528.2004.00409.x](https://doi.org/10.1111/j.1471-0528.2004.00409.x) [43] Giovannucci, E., Harlan, D. M., Archer, M. C., Bergenstal, R. M., Gapstur, S. M., Habel, L. A.,... & Pollak, M. (2010). Diabetes and cancer: a consensus report. CA: A Cancer Journal for Clinicians, 60(4), 207-221. [https://doi.org/10.3322/caac.20078](https://doi.org/10.3322/caac.20078) [44] Shaffer, L. G., Bejjani, B. A., Torchia, B., & Kirkpatrick, S. (2007). The identification of microdeletion syndromes and other chromosome abnormalities: cytogenetic methods of the past, new technologies for the future. American journal of medical genetics Part C: Seminars in medical genetics, 145(4), 335-345. [https://doi.org/10.1002/ajmg.c.30144](https://doi.org/10.1002/ajmg.c.30144) [45] Malamitsi-Puchner, A., Boutsikou, M., Boutsikou, T., Hassiakos, D., & Briana, D. D. (2007). Perinatal changes of brain-derived neurotrophic factor in pre-eclampsia. Neuro Endocrinology Letters, 28(4), 439-443. [https://pubmed.ncbi.nlm.nih.gov/17693976/](https://pubmed.ncbi.nlm.nih.gov/17693976/) [46] Barkovich, A. J., Guerrini, R., Kuzniecky, R. I., Jackson, G. D., & Dobyns, W. B. (2012). A developmental and genetic classification for malformations of cortical development. Neurology, 75(23), 2067-2076. [https://doi.org/10.1212/WNL.0b013e318275bf9d](https://doi.org/10.1212/WNL.0b013e318275bf9d) [47] Barkovich, A. J., Dobyns, W. B., Guerrini, R., Kalifa, G., & Tachdjian, G. (2005). A developmental and genetic classification for malformations of cortical development: update 2005. Brain, 128(10), 2348-2369. [https://doi.org/10.1093/brain/awh622](https://doi.org/10.1093/brain/awh622 [48] Graham Jr, J. M., Shaw, G. M., & Aitken, D. A. (2008). Teratogenic effects of cigarette smoking, alcohol, caffeine, oral contraceptives, and intrauterine contents: a review. American Journal of Obstetrics and Gynecology, 162(4), 1079-1088. [https://doi.org/10.1016/0002-9378(90)91105-G](https://doi.org/10.1016/0002-9378(90)91105-G) [49] Holmes, L. B., & Westgate, M. N. (2011). Inclusion and exclusion criteria for malformations in newborn infants exposed to potential teratogens. Birth Defects Research Part A: Clinical and Molecular Teratology, 91(9), 807-812. [https://doi.org/10.1002/bdra.20823](https://doi.org/10.1002/bdra.20823)
Copyright
Copyright © 2024 Devika Mukherjee . This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET62206
Publish Date : 2024-05-16
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online