

Ijraset Journal For Research in Applied Science and Engineering Technology
Machine Learning in Genomic Data Analysis for Personalized Medicine
Authors: Aayushi Saraswat, Shrikanth Roopesh
DOI Link: https://doi.org/10.22214/ijraset.2024.63975
Certificate: View Certificate
Abstract
The advent of precision medicine marks a transformative shift from traditional symptom-based treatments to more personalized approaches that account for individual genetic variations. Central to this evolution is the integration of genomic data, which offers profound insights into the biological underpinnings of disease and enables the development of targeted therapies. Machine learning (ML) has emerged as a pivotal tool in this domain, capable of deciphering complex patterns in high-dimensional genomic data, thereby enhancing early disease risk prediction and informing tailored therapeutic strategies. This review explores the intersection of machine learning and genomic medicine, highlighting how advanced computational techniques, including deep learning and other ML approaches, are driving innovation in personalized healthcare. By examining the application of these technologies in areas such as disease risk assessment, precision oncology, and pharmacogenomics, this paper elucidates the current state of the field and identifies future directions for research. Ethical considerations, such as data privacy, model transparency, and bias mitigation, are also discussed, emphasizing the need for responsible and equitable implementation of ML in clinical practice. Through this analysis, the paper aims to underscore the potential of machine learning to revolutionize personalized medicine and improve patient outcomes.
Introduction
I. INTRODUCTION
In recent years, the fields of precision medicine and genomic medicine have emerged as pivotal forces in the transformation of healthcare, offering new opportunities to enhance the diagnosis, treatment, and prevention of diseases. Precision medicine, a concept built upon the understanding of human genetics, environmental factors, and individual lifestyles, aims to tailor medical interventions to the unique characteristics of each patient. This approach represents a fundamental shift from the traditional, symptom-driven model of medical practice to a more individualized, data-driven strategy that holds the promise of more effective treatments, optimized healthcare expenditure, and improved patient outcomes. Medical error ranks as the third leading cause of death, following heart failure and cancer. Recent studies estimate that approximately 180,000 to 251,000 individuals in the USA die annually due to errors in medical reports.[1] The impact of precision medicine is already being felt in various domains, including oncology, cardiovascular disease, and chronic inflammatory conditions, where customized therapies are leading to significant improvements in patient survival rates and quality of life.
Simultaneously, genomic medicine, a relatively newer but rapidly growing discipline, focuses on the application of genetic information to guide clinical decision-making. A genome acts as the blueprint for building an organism. It has been known since 1953 that DNA molecules are the physical carriers of genetic information, and by 2001, the Human Genome Project had produced a draft of the typical human genome's raw information [2]-[4]. By leveraging the genetic makeup of individuals, genomic medicine has the potential to revolutionize the way diseases are diagnosed and treated, particularly in areas such as oncology, rare genetic disorders, infectious diseases, and pharmacology. The integration of genomic data into clinical practice enables healthcare providers to identify disease-causing mutations with greater precision, thereby allowing for the development of targeted therapies that are specifically tailored to the genetic profiles of individual patients. This personalized approach not only enhances the effectiveness of treatments but also minimizes the risk of adverse reactions, paving the way for more precise and safer healthcare interventions.
However, the implementation of precision and genomic medicine is not without its challenges. The increasing complexity of medical data, coupled with the sheer volume of genomic information being generated, presents significant obstacles for healthcare providers. Traditional methods of data analysis are often insufficient to manage and interpret this vast amount of information, leading to concerns about medical errors, which remain a leading cause of mortality worldwide. In this context, the advent of artificial intelligence (AI) and machine learning (ML) offers a promising solution to these challenges. AI, with its ability to mimic human intelligence and process large datasets, and ML, a subset of AI focused on pattern recognition and predictive analytics, are becoming indispensable tools in the analysis of genomic and clinical data.
AI is currently being used to automate the processing of data from various sources, summarize electronic health records (EHRs) and handwritten physician notes, integrate health records, and manage data on a cloud scale [5]-[10].
Machine learning, in particular, has shown tremendous potential in enhancing the accuracy and effectiveness of genomic medicine. By employing sophisticated algorithms and deep learning techniques, ML models can integrate and analyze diverse datasets—ranging from clinical records and genomic sequences to metabolomics and imaging data. These models can identify complex patterns and relationships that are not immediately apparent to human clinicians, enabling early disease detection, accurate risk prediction, and the development of personalized treatment strategies. Moreover, the application of ML in genomic medicine is not limited to data analysis; it also plays a crucial role in the discovery of new biomarkers, the identification of potential therapeutic targets, and the development of predictive models for various diseases.
In recent years, personalized medicine has emerged as a pivotal innovation in health-related research, offering significant potential for enhancing patient care. [11,12]. The integration of machine learning into precision and genomic medicine is transforming the healthcare landscape, ushering in a new era of personalized medicine that holds the potential to improve patient outcomes and reduce healthcare costs. However, despite the significant advancements in this field, several challenges remain. These include the need for more robust computational models, the integration of heterogeneous data sources, and the development of standardized frameworks for the application of ML in clinical settings. As research in this area continues to evolve, addressing these challenges will be critical to fully realizing the potential of ML in genomic medicine.
This review paper seeks to explore the intersection of machine learning and genomic medicine, providing an in-depth analysis of how ML is being used to solve key problems in this domain. The paper will examine the current state of the art, highlighting recent advancements and the impact of ML on personalized medicine. It will also discuss the challenges that lie ahead, as well as potential future directions for research in this rapidly evolving field. By doing so, this paper aims to underscore the transformative potential of machine learning in advancing precision and genomic medicine, ultimately contributing to the goal of achieving truly personalized healthcare for all.
II. MACHINE LEARNING IN PRECISION MEDICINE
Machine learning (ML) has emerged as a pivotal component of artificial intelligence (AI), providing sophisticated computational models that recognize and interpret patterns within extensive datasets. First introduced by Arthur Samuel in the 1950s, the concept of "machine learning" has evolved considerably, leading to significant advancements in the field. ML encompasses various methodologies, primarily categorized into supervised learning, unsupervised learning, and reinforcement learning. [13]
Supervised learning, one of the most widely used approaches, includes both classification and regression techniques. Classification involves predicting discrete, categorical outcomes based on labeled training data, such as diagnosing malignancy from biopsy samples. In contrast, regression focuses on forecasting continuous numeric outcomes, exemplified by predicting the interval between a patient’s hospital discharge and potential readmission. These techniques enable precise predictions and classifications, which are essential in precision medicine.
Unsupervised learning, another critical category, includes clustering methods that segment data into groups without predefined labels. This approach is particularly useful for discovering hidden patterns within data, such as determining the prevalence of diseases in populations exposed to environmental risks. Unlike supervised learning, clustering does not rely on labeled data, making it valuable for exploratory data analysis and pattern recognition.
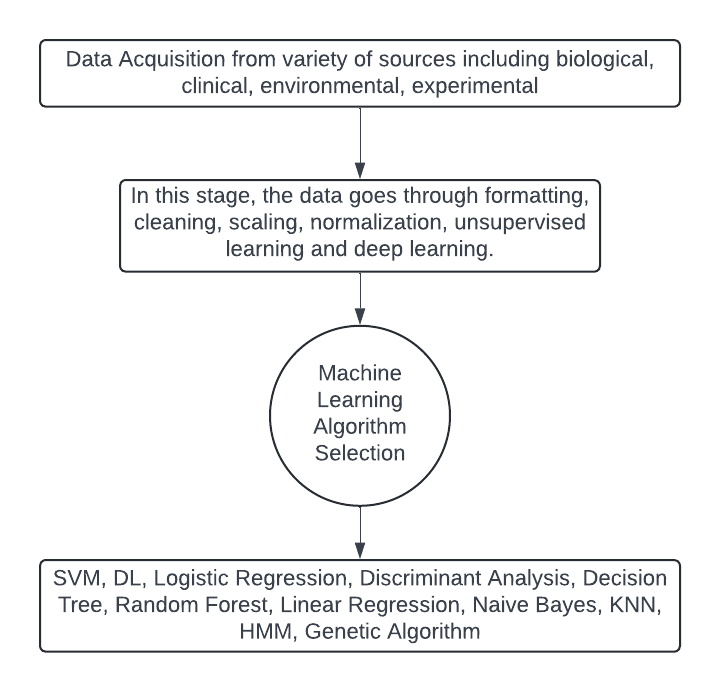
Fig 1. Machine learning process
Reinforcement learning, while less prevalent in precision medicine compared to supervised and unsupervised methods, involves training models through a system of rewards and punishments. Positive feedback encourages the model to repeat beneficial actions, while negative feedback guides it to avoid detrimental decisions. Although reinforcement learning plays a minor role in precision medicine, it remains a valuable tool in scenarios requiring adaptive and iterative learning.
Machine learning is revolutionizing healthcare by enhancing various aspects of patient care and clinical decision-making. Its applications include continuous patient monitoring to detect health changes, analyzing disease patterns to inform treatment strategies, and aiding in accurate diagnosis and personalized medication prescriptions. Additionally, ML contributes to patient-centered care by tailoring interventions to individual needs, reducing clinical errors through data-driven support, and predicting high-risk emergencies such as sepsis.
In the realm of precision medicine, several machine learning algorithms play crucial roles in analyzing complex datasets and enhancing clinical decision-making. Here, we discuss some of the key algorithms and their specific contributions to the field.
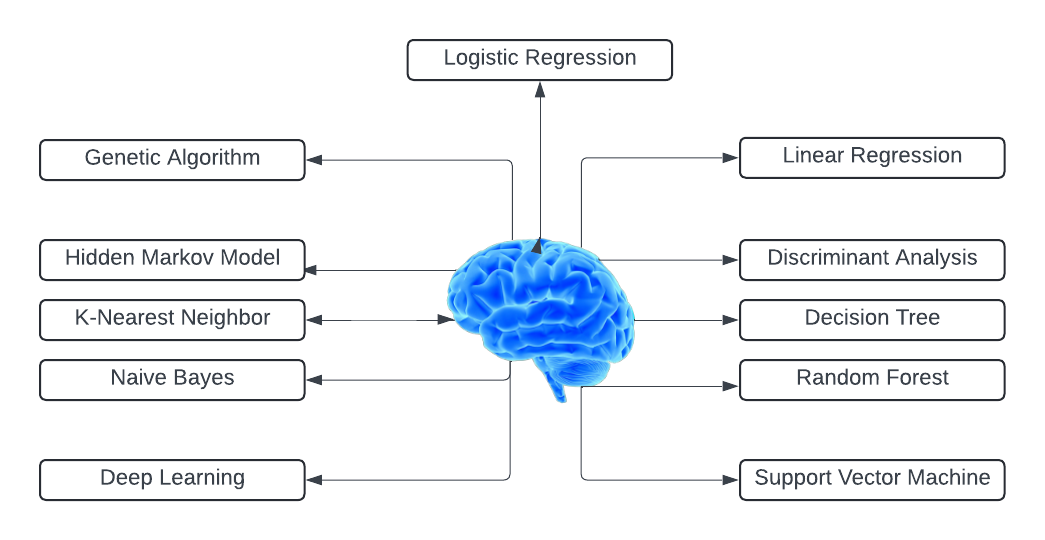
Fig. 2 Types of machine learning algorithms
A. Support Vector Machines (SVM)
Support Vector Machines (SVM) are supervised learning models employed for tasks involving classification and regression. SVMs work by finding the optimal hyperplane that separates data points of different classes with the maximum margin. In precision medicine, SVMs are employed for classifying disease states and predicting patient outcomes. For example, SVMs can differentiate between benign and malignant tumors based on histopathological features, aiding in cancer diagnosis. It processes clinical, molecular, and genomic data to validate oral cancer diagnoses and identify mental health conditions. [14]-[16]. Their ability to handle high-dimensional data makes them suitable for genomic data analysis, where they can identify biomarkers and predict disease susceptibility.
B. Genetic Algorithms
Genetic Algorithms (GAs) are optimization methods modeled after the concepts of natural selection. They use processes such as mutation, crossover, and selection to evolve solutions to problems. In precision medicine, GAs are used for feature selection and parameter optimization in predictive models. For instance, GAs can identify the most relevant genetic markers associated with a disease, helping to refine diagnostic tools and treatment plans. Their adaptability allows them to handle complex and nonlinear relationships in medical data.
C. Hidden Markov Model (HMM)
Hidden Markov Models (HMM) are statistical models that represent systems with hidden states. They are particularly useful for sequence data, where observations are assumed to be generated by a sequence of hidden states. In precision medicine, HMMs are applied to analyze genetic sequences and gene expression data.
They can model the progression of diseases such as cancer by capturing the temporal dynamics of gene expression changes. HMMs are also used in identifying genetic mutations and understanding their impact on disease development. The Hidden Markov Model (HMM) algorithm has been applied across various medical fields, with notable real-time contributions including the extraction of drug side effects from online healthcare forums, reduction of healthcare costs, analysis of personal health check-up data, observation of circadian patterns in telemetric activity data, clustering and modeling patient journeys in medical settings, scrutiny of healthcare service utilization post-injury through transport systems, analysis of infant cry signals, and prediction of individuals entering countries with significant numbers of asynchronies.[17]-[23]
D. Linear Regression
Linear Regression is a statistical approach used to model the relationship between a dependent variable and one or more independent variables. It is widely used for predicting continuous outcomes. In precision medicine, linear regression models can predict disease progression and treatment response based on clinical and genomic data. For example, linear regression can estimate the likelihood of disease recurrence based on patient characteristics and treatment history, guiding personalized treatment strategies.
E. Discriminant Analysis (DA)
Discriminant Analysis (DA) is a classification technique that seeks to find a linear combination of features that best separates different classes. DA is useful in scenarios where the goal is to classify patients into distinct diagnostic categories. In precision medicine, DA can be used to identify disease subtypes and predict patient outcomes based on clinical and genetic information. It helps in stratifying patients into different risk groups, which is essential for tailoring personalized treatment approaches.
F. Decision Tree
Decision Trees are hierarchical models that use a tree-like structure to make decisions based on input features. Each internal node represents a decision based on a feature, and each leaf node represents a classification or prediction. In precision medicine, decision trees are used for diagnosing diseases, predicting patient responses to treatments, and identifying key risk factors. Their interpretability allows clinicians to understand the decision-making process and apply it to patient care.
G. Logistic Regression
Logistic Regression is a statistical technique applied to binary classification problems, modeling the probability of a binary outcome based on one or more predictor variables. In precision medicine, logistic regression is applied to predict the likelihood of disease occurrence or response to treatment. For example, logistic regression can estimate the probability of a patient developing a specific condition based on genetic and clinical data, supporting early diagnosis and preventive measures.
H. Naïve Bayes
Naïve Bayes is a probabilistic classifier based on Bayes' theorem, which assumes independence between features. Despite this simplification, it performs well in many classification tasks. In precision medicine, Naïve Bayes classifiers are used for disease prediction and risk assessment. This algorithm is applied across various medical fields, including predicting risks associated with Mucopolysaccharidosis type II, analyzing censored and time-to-event data, classifying electronic health records (EHR), enhancing clinical diagnosis through decision support, extracting genome-wide data to identify Alzheimer's disease, modeling decisions related to cardiovascular conditions, assessing the quality of healthcare services, and developing predictive models for cancers of the brain, asthma, prostate, and breast.[24]-[33]
I. Deep Learning Models
Deep Learning Models are a subset of machine learning that uses neural networks with multiple layers to model complex patterns in data. In precision medicine, deep learning is applied to various tasks, including image analysis (e.g., radiology and pathology images), genomics, and drug discovery. These models excel at handling large-scale data and extracting features that are not easily captured by traditional methods. They have shown promise in improving diagnostic accuracy and predicting treatment responses.
J. Random Forest
Random Forest is an ensemble learning method that combines multiple decision trees to improve classification and regression performance. By averaging the predictions of several trees, Random Forest reduces overfitting and increases model robustness. In precision medicine, Random Forest is used for feature selection, disease classification, and prediction of patient outcomes. Its ability to handle large datasets and complex interactions makes it valuable for analyzing genomic and clinical data.
K. K- Nearest Neighbor
K-Nearest Neighbor (KNN) is a simple, instance-based learning algorithm that classifies data points based on their proximity to labeled examples. In precision medicine, KNN is used for patient classification and disease prediction. For instance, KNN can identify patients with similar genetic profiles and predict disease risks based on the outcomes of similar cases. Its simplicity and effectiveness in handling diverse data types make it a useful tool in clinical settings. It has been used to safeguard confidential clinical prediction data in e-Health clouds, classify patterns for breast cancer diagnosis, predict pancreatic cancer through published research, model diagnostic performance, detect gastric cancer, classify patterns for health monitoring applications, categorize medical datasets, and manage electronic health record (EHR) data. [34]-[39]
III. OVERVIEW OF GENOMIC DATA
Genomic data serves as the foundation for understanding the intricate molecular mechanisms that drive biological processes and influence the development of various diseases. By providing a comprehensive view of an organism's genetic material, genomic data allows researchers to investigate the relationship between genes and phenotypes, identify genetic variants associated with diseases, and explore potential therapeutic targets. The rapid advancements in sequencing technologies over the past two decades have led to an explosion of genomic data, offering unprecedented opportunities to unravel the complexities of the genome.
The analysis of genomic data, however, is not without its challenges. The vast amount of information generated from sequencing experiments, coupled with the inherent complexity of biological systems, necessitates the development of sophisticated computational methods to interpret the data accurately. Additionally, the variability in data quality, arising from technical limitations, noise, and biological heterogeneity, poses significant obstacles to drawing meaningful conclusions from genomic studies.
This section provides an overview of the different types of genomic data commonly used in research, including DNA sequencing data, transcriptomic data, and epigenomic data. Each type of data offers unique insights into the genome's structure and function, contributing to a holistic understanding of gene regulation and its impact on health and disease. Furthermore, the section discusses the key challenges encountered in genomic data analysis, emphasizing the need for advanced methodologies to address issues related to high dimensionality, noise, missing data, and data heterogeneity. By navigating these challenges, researchers can fully leverage the potential of genomic data to drive discoveries in genomics and precision medicine.
A. Types of Genomic Data
Genomic data encompasses a broad range of information types, each providing unique insights into the structure, function, and regulation of the genome. The primary types of genomic data include DNA sequencing data, transcriptomic data, and epigenomic data. These data types form the cornerstone of genomics research, allowing scientists to explore the complex interactions between genes and their environment, and to understand how these interactions contribute to various biological processes and disease states. In genomics and biology, there is a growing recognition that investing in new computational techniques might be more beneficial than solely focusing on data collection—a point that computational biologists have long advocated. As highlighted by critics of the data-centric approach. For example, despite nearly $1 billion spent on The Cancer Genome Atlas (TCGA) project, there is ongoing debate about whether the focus should remain on sequencing or shift towards analyzing the functional aspects of the data [40]. Computer systems capable of analyzing genomic text offer numerous applications in genomic medicine. A notable advancement in this field is the development of "gene editing" technologies, which enable scientists to modify the genomes of living cells with unprecedented precision. These technologies allow for targeted interventions, such as removing harmful mutations or inserting new sequences at specific locations within a genome. As gene editing technologies evolve, it becomes increasingly crucial to predict the outcomes of these modifications through computational models. Understanding how to implement these edits is not sufficient; it is equally important to anticipate their effects in silico [41,42].
1) DNA Sequencing Data: Whole Genome Sequencing (WGS): Whole Genome Sequencing is a comprehensive method for analyzing the entire genetic makeup of an organism. WGS provides a complete snapshot of the genome, including all coding (exonic) and non-coding (intronic, regulatory) regions, repetitive elements, structural variants, and single nucleotide polymorphisms (SNPs). By sequencing the entire genome, WGS offers an unparalleled level of detail, enabling researchers to identify both common and rare genetic variants associated with disease, as well as to study the evolutionary history of genomes. However, the sheer volume of data generated by WGS presents significant computational and analytical challenges, requiring powerful algorithms to process and interpret the data effectively. Exome Sequencing: Exome sequencing is a targeted approach that focuses on sequencing only the exonic regions of the genome, which constitute about 1-2% of the entire genome. Despite covering a smaller portion of the genome compared to WGS, exome sequencing is highly valuable because it captures the regions that directly encode proteins, which are often where disease-causing mutations are found. Exome sequencing is widely used in clinical settings to identify genetic variants responsible for Mendelian disorders and to discover novel disease genes. Due to its cost-effectiveness relative to WGS, exome sequencing is a popular choice for studies where the focus is on protein-coding regions, although it may miss important regulatory variants located outside these regions.
2) Transcriptomic Data: RNA Sequencing (RNA-Seq): RNA sequencing is a powerful technique for profiling the transcriptome—the complete set of RNA transcripts produced by the genome under specific conditions. RNA-Seq provides quantitative data on gene expression levels, alternative splicing events, and post-transcriptional modifications, allowing researchers to study how genes are regulated and how they respond to different stimuli. RNA-Seq is particularly valuable for identifying differential gene expression between normal and diseased tissues, uncovering novel transcripts, and studying the complexity of the transcriptome at a single-cell level. The depth of information provided by RNA-Seq makes it a crucial tool for understanding the functional consequences of genetic variation and for discovering biomarkers that can be used in diagnosis and treatment. Single-Cell RNA Sequencing (scRNA-Seq): Single-cell RNA sequencing takes the analysis of transcriptomic data a step further by allowing the study of gene expression at the level of individual cells. This technique has revolutionized our understanding of cellular heterogeneity, revealing the diversity of cell types and states within complex tissues. scRNA-Seq is instrumental in identifying rare cell populations, mapping developmental trajectories, and understanding the cellular basis of diseases such as cancer. The ability to analyze gene expression on a cell-by-cell basis provides unprecedented insights into how genetic and environmental factors influence cellular function and behavior, making scRNA-Seq a powerful tool in both basic and translational research.
3) Epigenomic Data: DNA Methylation Data: DNA methylation is one of the most studied epigenetic modifications, involving the addition of a methyl group to the cytosine residues in DNA, primarily at CpG dinucleotides. Methylation patterns play a crucial role in regulating gene expression, with hypermethylation often leading to gene silencing and hypomethylation associated with gene activation. DNA methylation data is essential for understanding how epigenetic changes contribute to gene regulation, development, and disease. Techniques such as bisulfite sequencing allow researchers to map DNA methylation patterns across the genome, providing insights into the epigenetic mechanisms underlying processes like imprinting, X-chromosome inactivation, and cancer progression.
Histone Modification Data: Histones are proteins around which DNA is wrapped to form chromatin, and their post-translational modifications (such as methylation, acetylation, and phosphorylation) play a key role in regulating chromatin structure and gene expression. Histone modification data provides information on the specific chemical modifications that occur on histone proteins, which in turn influence the accessibility of DNA to transcription factors and other regulatory proteins. Techniques like chromatin immunoprecipitation followed by sequencing (ChIP-Seq) are used to profile histone modifications across the genome, revealing the dynamic nature of chromatin and its impact on gene regulation. Understanding histone modifications is critical for elucidating the epigenetic control of gene expression and for identifying potential targets for epigenetic therapies.
Chromatin Accessibility Data (e.g., ATAC-Seq): Chromatin accessibility refers to the ease with which transcriptional machinery and other regulatory proteins can access DNA, which is influenced by the structure and composition of chromatin. Techniques like ATAC-Seq (Assay for Transposase-Accessible Chromatin using sequencing) allow researchers to map regions of open chromatin, providing insights into the regulatory elements that control gene expression. Chromatin accessibility data is essential for understanding how the genome is organized within the nucleus and how changes in chromatin structure can influence cellular function and identity. By identifying accessible regions of the genome, researchers can pinpoint active enhancers, promoters, and other regulatory elements that play key roles in controlling gene expression in different cell types and conditions.
Each type of genomic data offers a unique perspective on the genome, contributing to a more comprehensive understanding of the genetic and epigenetic mechanisms that drive biological processes. By integrating these diverse data types, researchers can gain deeper insights into the complex interplay between genes, regulatory elements, and environmental factors, paving the way for new discoveries in genomics and precision medicine.
B. Challenges in Genomic Data Analysis
The analysis of genomic data presents a range of complex challenges due to the intrinsic properties of the data and the biological systems they represent. These challenges arise from the high dimensionality and complexity of the data, the presence of noise and missing information, and the heterogeneity of the data sources. Understanding and addressing these challenges are crucial for extracting meaningful insights from genomic data and advancing our knowledge in fields such as genomics, precision medicine, and systems biology.
One of the most significant challenges in genomic data analysis is the high dimensionality of the data. Genomic datasets often consist of millions of variables (e.g., single nucleotide polymorphisms (SNPs), gene expression levels, methylation sites) but may contain relatively few samples. This phenomenon, known as the "curse of dimensionality," complicates the application of traditional statistical methods and machine learning algorithms, which are not always well-suited to handle such high-dimensional data.
- High Dimensionality: In genomics, the number of features (such as genetic variants) can vastly outnumber the number of samples available for analysis. This imbalance can lead to overfitting, where a model learns the noise in the training data rather than the underlying biological signals, resulting in poor generalization to new data. Dimensionality reduction techniques, such as principal component analysis (PCA) or t-distributed stochastic neighbor embedding (t-SNE), are often employed to mitigate this issue, but these methods may also result in the loss of potentially important information.
- Complexity of Biological Systems: The biological systems that genomic data represent are inherently complex, involving interactions between thousands of genes, proteins, and other molecules. These interactions occur within a dynamic environment, influenced by factors such as development, environmental conditions, and disease states. Capturing and modeling these complex interactions is a major challenge in genomic data analysis. Systems biology approaches, which aim to model the interactions within biological networks, can provide insights into the underlying mechanisms, but require sophisticated computational tools and large-scale data integration. Genomic data is often noisy and incomplete, which can obscure true biological signals and lead to incorrect conclusions if not properly handled. Noise can arise from various sources, including technical limitations of sequencing technologies, biological variability, and sample contamination. Missing data is also a common issue, particularly in large-scale studies, where not all variables may be measured or recorded for every sample.
- Noise in Sequencing Data: Next-generation sequencing (NGS) technologies, while powerful, are not without errors. Sequencing errors can introduce false variants (such as SNPs or indels) into the data, complicating downstream analyses. Additionally, biological noise, such as random fluctuations in gene expression levels, can further confound the interpretation of results. To address noise, bioinformaticians often employ techniques such as quality control filtering, error correction algorithms, and robust statistical methods that are less sensitive to outliers.
- Missing Data: Missing data can occur for various reasons, including technical failures (e.g., failed sequencing runs), limited sample availability, or incomplete data collection. Missingness can be problematic, particularly if it is not random, as it can introduce bias into the analysis. Imputation methods, which estimate missing values based on observed data, are commonly used to address this issue. However, the accuracy of imputation depends on the quality and completeness of the available data, and incorrect imputation can lead to misleading results. Genomic data is highly heterogeneous, encompassing different types of biological information (e.g., genetic variants, gene expression, epigenetic modifications) collected from diverse populations, tissues, and conditions. This heterogeneity poses significant challenges for data integration, comparison, and interpretation.
- Biological Heterogeneity: Biological heterogeneity refers to the variation in genomic features across different individuals, populations, and species, as well as within different tissues and cell types of the same organism. For example, genetic diversity between populations can result in population-specific genetic variants that complicate the interpretation of genome-wide association studies (GWAS). Similarly, tissue-specific gene expression patterns and epigenetic modifications add another layer of complexity to data analysis. Addressing biological heterogeneity often requires the development of models that can account for these differences, such as mixed-effect models or hierarchical Bayesian models.
- Technological Heterogeneity: Technological heterogeneity arises from the use of different platforms, protocols, and sequencing technologies to generate genomic data. For instance, data generated by different sequencing platforms (e.g., Illumina vs. Oxford Nanopore) may have varying levels of accuracy, coverage, and bias. Additionally, the choice of library preparation methods, sequencing depth, and bioinformatics pipelines can all introduce variability into the data. To mitigate these issues, researchers often standardize data processing pipelines and employ normalization techniques to make datasets more comparable.
- Contextual Heterogeneity: Contextual heterogeneity refers to the variability in experimental conditions, such as differences in sample collection times, environmental exposures, or disease states. This type of heterogeneity can lead to confounding effects, where observed associations between genomic features and phenotypes are driven by unaccounted-for variables. Careful experimental design, including the use of matched controls and longitudinal studies, can help to reduce the impact of contextual heterogeneity. Additionally, advanced statistical methods, such as covariate adjustment and causal inference models, can be used to account for confounding factors. As the volume of genomic data continues to grow, scalability and computational efficiency have become major concerns. Analyzing large-scale genomic datasets requires substantial computational resources, including high-performance computing clusters and cloud-based platforms. However, even with these resources, the sheer size and complexity of the data can make it difficult to perform timely analyses.
- Scalability: Traditional bioinformatics tools and algorithms may not scale well to handle the massive datasets generated by modern genomic studies. For example, alignment and variant calling algorithms that were developed for smaller datasets may become prohibitively slow or memory-intensive when applied to whole-genome sequencing data from thousands of individuals. To address this challenge, researchers are developing more efficient algorithms, parallel processing techniques, and distributed computing frameworks that can scale to larger datasets.
- Data Storage and Management: The storage and management of genomic data also present significant challenges. Genomic datasets can be terabytes or even petabytes in size, requiring substantial storage infrastructure. Additionally, the need to store raw sequencing data, processed data, and intermediate analysis results adds to the complexity of data management. Effective data compression techniques, along with well-organized data repositories and metadata standards, are essential for managing these large datasets.
- Data Privacy and Security: The sensitive nature of genomic data, which can reveal information about an individual's ancestry, health risks, and other personal traits, raises important privacy and security concerns. Ensuring the confidentiality and security of genomic data is critical, particularly in clinical settings where patient data is involved. Strategies such as data anonymization, encryption, and secure access controls are employed to protect genomic data, but these measures must be balanced with the need for data sharing and collaboration in research.
IV. INTEGRATION OF MACHINE LEARNING IN GENOMIC MEDICINE
The integration of machine learning (ML) into genomic medicine represents a significant advancement in the field of healthcare, revolutionizing how we interpret and utilize genetic information. Genes, the fundamental units of heredity, are estimated to number between 20,000 and 25,000 in humans. Each individual inherits two copies of each gene, one from each parent. The human genome comprises both coding genes, which include those that code for proteins and non-proteins. Genes can vary significantly in length, ranging from as few as a hundred to as many as two million DNA bases. Consequently, the genome reflects both the number of genes and the complexity of gene networks. As Mukherjee describes, "The human genome is fiercely innovative, dynamic, and exhibits a variety of characteristics including unexpected beauty, historical richness, inscrutability, vulnerability, resilience, adaptability, repetitiveness, and uniqueness" [43,44]. As the volume and complexity of genomic data grow exponentially, traditional analytical methods are often insufficient to extract meaningful insights. ML provides powerful tools to address these challenges by enabling the analysis of large and intricate datasets with greater accuracy and efficiency.
Machine learning algorithms excel in recognizing patterns and making predictions based on vast amounts of data, making them particularly well-suited for genomic medicine. These algorithms can process data from genome sequencing, phenotyping, and variant identification to uncover insights that might be missed through conventional methods. The application of ML in these areas enhances our ability to understand genetic influences on health, predict disease risks, and tailor treatments to individual patients.
The integration of ML into genomic medicine encompasses several critical areas. In genome sequencing, ML algorithms are used to analyze complex genomic data, improve sequencing accuracy, and identify genetic variants. In phenotyping, ML helps link genomic information with observable traits and clinical outcomes, enabling more precise predictions and personalized treatment plans.
For variant identification and interpretation, ML automates the process of detecting and annotating genetic variants, improving the efficiency and accuracy of genetic diagnostics.
As we explore these areas in detail, it becomes evident that ML is not just a supplementary tool but a fundamental component of modern genomic medicine, driving innovation and improving patient outcomes through its ability to handle and interpret complex genetic data.
A. Genome Sequencing
Genome sequencing is a foundational component of genomic medicine, providing comprehensive insights into the complete DNA sequence of an organism. Researchers have developed a machine learning model designed to predict DNA-binding rates from sequence data, which assists in the design of effective probes. Additionally, errors can arise from base calling in raw DNA sequencing data. To address this, several deep learning methods have been developed specifically for enhancing the accuracy of base calling with Oxford Nanopore long-read sequencers [45]-[47]. This process allows researchers and clinicians to decipher the genetic code, identify genetic variations, and understand their implications for health and disease. With the rapid advancement of sequencing technologies, the integration of machine learning (ML) has become increasingly important in enhancing the accuracy and utility of genome sequencing data.
Whole Genome Sequencing involves determining the complete nucleotide sequence of an organism's genome. This method provides a comprehensive view of both coding and non-coding regions, capturing all genetic variations, including single nucleotide polymorphisms (SNPs), insertions, deletions, and structural variants. WGS is instrumental in understanding complex genetic conditions, exploring genetic diversity, and identifying novel genetic markers for diseases.
Whole Exome Sequencing focuses on sequencing the exonic regions of the genome, which encode proteins. Although WES covers only about 1-2% of the genome, it is particularly useful for identifying mutations that affect protein function. WES is often employed in research and clinical settings to pinpoint genetic causes of rare and inherited diseases, as many disease-causing mutations occur in the exonic regions.
Targeted Sequencing involves sequencing specific regions of interest within the genome, such as genes associated with particular diseases or pathways. This approach is cost-effective and provides high coverage of the targeted regions, making it valuable for both research and clinical diagnostics. Targeted panels are used for conditions with well-defined genetic markers, such as certain types of cancer.
Sequencing methods involve isolating short DNA or RNA fragments, typically around 100 base pairs in length, which are bound to the protein of interest. These fragments are then sequenced and mapped to a reference genome. The principle behind this approach is that areas where the mapped reads overlap and accumulate, forming a "peak," indicate regions where the protein preferentially binds. Common experimental protocols for studying DNA-binding proteins include ChIP-seq, while methods for RNA-binding proteins include RIP-seq and CLIP-seq.[48]
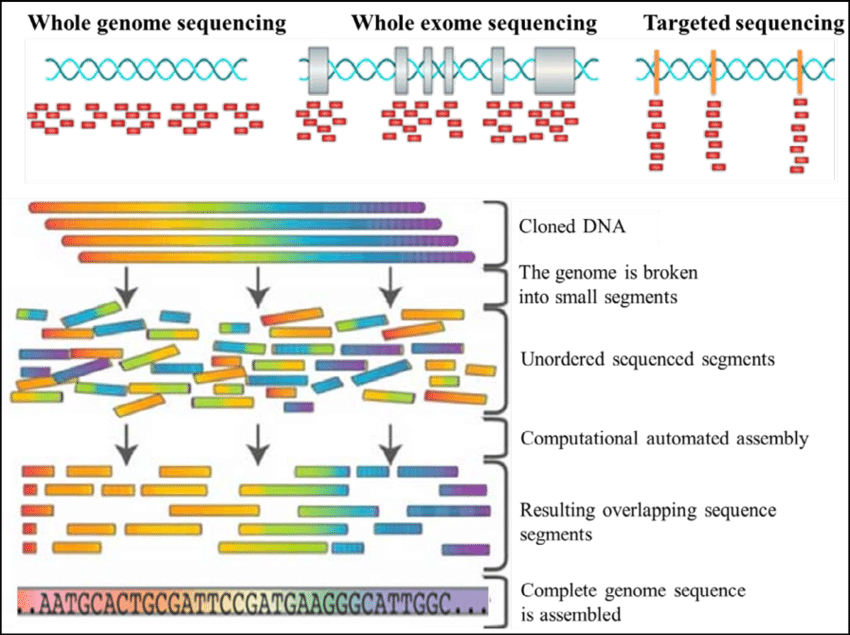
Fig.3 Types of genome sequencing
The data processing and analysis of sequencing data involve several critical steps, including quality control, alignment, and variant calling. Raw sequencing reads are first assessed for quality, and errors are corrected before aligning the reads to a reference genome, where variants are identified based on differences between the sample and the reference. Machine learning algorithms can enhance these steps by improving read alignment accuracy and detecting subtle variations. Following variant calling, the next crucial step is interpreting the identified variants, which involves classifying them based on their potential impact on gene function and their association with diseases. Here, ML algorithms can assist in predicting the pathogenicity of variants by integrating various sources of information, such as functional annotations, population frequency, and evolutionary conservation.
The vast volume and complexity of data generated by genome sequencing pose significant challenges in storage, processing, and analysis. Machine learning techniques are essential for managing this complexity, enabling efficient data handling and the extraction of actionable insights from intricate genomic data. Furthermore, interpreting rare genetic variants presents additional challenges due to limited population data and functional knowledge. Machine learning models can improve the interpretation of rare variants by leveraging large-scale datasets, incorporating diverse types of genomic and clinical information, and applying sophisticated algorithms to predict variant effects and their associations with diseases. Integrating genomic data with clinical information is crucial for translating sequencing results into actionable medical insights. Machine learning can facilitate this integration by aligning genomic findings with clinical outcomes, predicting patient responses to treatments, and supporting personalized medicine approaches.
Looking to the future, ongoing advancements in machine learning algorithms and sequencing technologies are expected to further enhance the accuracy and speed of genome sequencing. Innovations such as real-time sequencing and improved ML models for data analysis will contribute to more precise and timely genomic insights. As genome sequencing becomes more accessible, machine learning-driven approaches will play a crucial role in personalized medicine, enabling tailored treatment strategies based on individual genetic profiles. This shift toward personalized care will be driven by advances in both sequencing technologies and machine learning algorithms. Additionally, the integration of multi-omics data, such as combining genomic data with other omics data (e.g., transcriptomics, proteomics), will provide a more comprehensive understanding of biological systems. Machine learning algorithms will be essential for integrating these diverse data types and extracting meaningful insights for both research and clinical applications.
B. Phenotyping
Phenotyping is the process of characterizing the observable traits or characteristics of an organism, which result from the interaction of its genotype with the environment. Machine learning approaches are being developed to extract phenotypic information from electronic health records (EHRs), improve the classification of phenotypes, and facilitate the analysis of phenotype data.[49,50] In the context of genomic medicine, phenotyping involves identifying and measuring traits that are relevant to understanding genetic influences on health and disease. The integration of machine learning (ML) has transformed phenotyping by enhancing the precision, scalability, and depth of phenotypic analyses.
- Types of Phenotypes: Clinical phenotypes refer to traits and conditions that are directly observable and measurable in patients, such as physical characteristics, disease symptoms, and health status. These phenotypes are often recorded through clinical assessments, medical histories, and diagnostic tests. For example, clinical phenotyping might involve identifying symptoms of a genetic disorder like cystic fibrosis or characterizing the progression of a disease such as cancer. Molecular phenotypes are traits at the molecular level, including gene expression levels, protein concentrations, and metabolite profiles. These phenotypes provide insights into the biochemical and cellular processes underlying genetic variations. Molecular phenotyping is often performed using techniques such as RNA sequencing for gene expression profiling, mass spectrometry for proteomics, and metabolomics assays for metabolic profiling. Imaging phenotypes involve the use of various imaging technologies to capture and analyze anatomical and functional features of organisms. Techniques such as MRI, CT scans, and PET scans are used to generate imaging data that can reveal structural abnormalities, functional changes, and disease progression. Imaging phenotyping is crucial for understanding the impact of genetic variations on physical structures and functions.
- Phenotyping Methods: Clinical assessments involve gathering detailed patient information through physical examinations, interviews, and standardized tests. These assessments help in identifying phenotypic traits relevant to genetic conditions and tracking changes over time. Clinical phenotyping often includes the use of diagnostic criteria, scales, and questionnaires to evaluate symptoms and disease severity.
High-throughput omics technologies, such as genomics, transcriptomics, proteomics, and metabolomics, enable comprehensive phenotyping at the molecular level. These technologies generate large-scale data sets that capture a wide range of molecular features. Machine learning algorithms are employed to analyze these data sets, identify patterns, and associate molecular phenotypes with genetic variations.
Imaging techniques provide detailed visual information about anatomical and functional aspects of organisms. Machine learning approaches are increasingly used to analyze imaging data, detect subtle changes, and classify phenotypic features. Techniques such as automated image analysis and deep learning models enhance the accuracy and efficiency of imaging phenotyping.
Machine learning (ML) plays a crucial role in integrating and analyzing diverse types of phenotypic data, including clinical, molecular, and imaging data. ML models are adept at identifying complex patterns within these data sets, correlating phenotypic traits with genetic variations, and uncovering new insights into disease mechanisms. Techniques such as feature selection, dimensionality reduction, and clustering are employed to manage large and complex phenotypic data sets, allowing for more nuanced and comprehensive analyses.
In the realm of predictive modeling, ML algorithms are used to forecast phenotypic outcomes based on genetic and environmental factors. These predictive models can estimate the risk of developing diseases, anticipate disease progression, and identify potential therapeutic targets. Supervised learning methods, such as classification and regression, are particularly effective in building these models, utilizing phenotypic data to predict outcomes with a high degree of accuracy.
Moreover, machine learning techniques are integral to phenotype-gene association studies, where the goal is to identify associations between phenotypes and genetic variants. Such studies aim to uncover genetic factors that influence phenotypic traits and contribute to disease susceptibility. ML algorithms enhance traditional methods like association testing, genome-wide association studies (GWAS), and pathway analysis, thereby improving the identification and understanding of phenotype-gene relationships.
Despite the advancements, several challenges persist in the application of ML to phenotyping. One major challenge is the heterogeneity of phenotypic data, as integrating data from diverse sources often leads to issues related to standardization, quality, and compatibility. Addressing these challenges requires the development of ML methods capable of harmonizing and normalizing data across different platforms and studies, ensuring consistency and reliability in the analyses.
Another significant challenge is the interpretability of ML models. While these algorithms can uncover intricate patterns within phenotypic data, interpreting the results and understanding their biological significance can be difficult. There is a growing need to enhance the interpretability of ML models, translating their findings into actionable insights that can be effectively applied in clinical practice.
Looking ahead, advances in ML have the potential to drive personalized phenotyping approaches. This involves tailoring assessments and analyses to individual patients based on their unique genetic and environmental profiles. Such personalized phenotyping will enhance the precision of diagnostics and treatment strategies, aligning with the broader goals of precision medicine. As ML continues to evolve, its application in phenotyping will likely lead to more accurate, personalized, and effective healthcare solutions.
C. Variant
Variant identification and interpretation are crucial components of genomic medicine, as they involve detecting genetic variations and understanding their implications for health and disease. Variants can range from single nucleotide changes to larger structural alterations, and their accurate identification and interpretation are essential for personalized medicine. Machine learning (ML) has increasingly been applied to enhance these processes, offering new capabilities in precision and efficiency. Numerous machine learning processes have been employed to enhance the specificity of detecting genuine somatic variations. Currently, deep learning methods are also being developed to advance this capability. By learning from training data, these techniques can more accurately differentiate true variant calls from artifacts caused by sequencing errors, coverage biases, or cross-contamination. A particularly challenging subset of variants is copy number variations (CNVs), which involve deletions or duplications of DNA segments. Machine learning strategies have been applied to improve the detection of CNVs with higher precision compared to individual CNV callers. This improvement is achieved by learning genomic features from a curated subset of verified CNVs and integrating data from multiple CNV detection algorithms.[51]-[55]
- Types of Variants: Single Nucleotide Polymorphisms (SNPs) are the most common type of genetic variation, involving a change of a single nucleotide in the DNA sequence. While many SNPs are benign and do not affect health, some can be associated with disease susceptibility or drug response. Identifying pathogenic SNPs and understanding their roles in disease mechanisms are key areas of research.
Insertions and Deletions (Indels) are variations where nucleotides are either inserted or deleted from the genome. These changes can impact gene function by altering coding sequences or regulatory regions. Indels can be challenging to detect and interpret due to their potential effects on the reading frame and gene expression.
Copy Number Variations (CNVs) involve changes in the number of copies of specific regions of the genome. They can range from small deletions or duplications to larger structural changes. CNVs are associated with various genetic disorders, including developmental and neuropsychiatric conditions.
Structural variants include larger-scale changes such as inversions, translocations, and duplications of genomic regions. These variants can disrupt gene function and contribute to complex diseases, including cancer. Accurate detection and interpretation of structural variants require sophisticated analytical techniques.
Advanced sequencing technologies, such as Whole Genome Sequencing (WGS) and Whole Exome Sequencing (WES), provide comprehensive data on genetic variants. WGS captures variations across the entire genome, while WES focuses on coding regions. These technologies generate vast amounts of data, requiring robust computational tools for variant identification. Variant calling algorithms analyze sequencing data to identify genetic variants. Tools such as GATK (Genome Analysis Toolkit), Samtools, and VarScan are commonly used for detecting SNPs and indels. For structural variants and CNVs, tools like DELLY and Lumpy are employed. Machine learning algorithms are increasingly used to improve the accuracy and sensitivity of variant calling by learning from large datasets and refining detection methods.
Once variants are identified, they need to be annotated and filtered to determine their potential clinical significance. Annotation involves adding information about the variant, such as its impact on gene function or its association with known diseases. Tools like ANNOVAR and VEP (Variant Effect Predictor) are used for this purpose. Machine learning approaches can enhance annotation by integrating data from multiple sources and predicting the functional impact of variants.
Interpreting the clinical significance of genetic variants involves assessing whether a variant is pathogenic, benign, or of uncertain significance. This process requires evaluating the variant's impact on gene function, its frequency in the population, and its association with disease. Resources such as ClinVar and the Human Gene Mutation Database (HGMD) provide curated information on variant-disease associations.
Understanding the functional impact of a variant involves determining how it affects the biological processes and pathways associated with a gene. Machine learning models can predict the effects of variants on protein structure and function, gene expression, and cellular processes. For example, tools like PolyPhen-2 and SIFT predict the impact of amino acid changes on protein function.
Machine learning models are increasingly used to predict the pathogenicity of genetic variants based on various features, such as sequence conservation, predicted functional impact, and population frequency. Models such as MutPred and CADD (Combined Annotation Dependent Depletion) integrate multiple sources of information to provide a comprehensive assessment of variant significance.
V. APPLICATIONS IN PERSONALIZED MEDICINE
The integration of machine learning (ML) into personalized medicine represents a paradigm shift in how healthcare is approached, emphasizing the customization of medical treatment to the individual characteristics of each patient. By leveraging advanced algorithms and vast amounts of data, ML enables the development of highly tailored interventions, optimizing both the efficacy and safety of medical treatments. This section provides an overview of the key applications of ML in personalized medicine, focusing on its transformative impact on various domains.
A. Precision Oncology
Precision oncology leverages machine learning (ML) and genomic data to customize cancer treatment based on individual patient profiles. The precision oncology approach necessitates the identification of a panel of biomarkers associated with therapeutic responses. Machine learning-based computational models are being developed to predict drug responses by utilizing multi-omics data and identifying response-predictive biomarkers [56]. Coudray et al. employed convolutional neural networks (CNNs) to accurately and thoroughly diagnose subtypes of lung cancer, including squamous cell carcinoma (LUSC) and adenocarcinoma (LUAD), as well as normal lung tissue, by analyzing digital scans from The Cancer Genome Atlas [57]. Huttunen et al. utilized automated classification techniques to categorize microscopy images of ovarian tissue obtained through multiphoton fluorescence imaging [58]. They also noted that their predictions were on par with those made by pathologists.
Similarly, Brinker et al. employed Convolutional Neural Networks (CNN) to automate the classification of dermoscopic melanoma images and discovered that it surpassed the diagnostic performance of both board-certified and junior dermatologists [59]. This approach aims to optimize therapeutic strategies according to the unique genetic and molecular characteristics of each patient’s tumor, enhancing treatment effectiveness and reducing side effects.
IBM Watson for Oncology employs advanced natural language processing and ML algorithms to analyze extensive medical literature, clinical trial results, and patient records. By integrating these data sources, Watson assists oncologists in identifying the most appropriate treatment options tailored to the genetic and molecular features of a patient's tumor. For instance, in a 2016 study, Watson for Oncology provided a recommendation for a patient with a rare lung cancer type, suggesting a targeted therapy based on an in-depth analysis of genetic mutations. This recommendation led to a positive treatment outcome, demonstrating the tool's capability to guide complex decision-making processes in oncology.
Tempus combines ML with genomic sequencing and clinical data to facilitate personalized cancer care. The Tempus platform evaluates tumor genomic profiles alongside patient medical histories to uncover actionable insights that inform treatment choices. For example, a patient with breast cancer was analyzed using Tempus’s platform, which identified a genetic mutation linked to drug resistance. Tempus recommended an alternative targeted therapy, resulting in significant tumor reduction and improved patient health.
Foundation Medicine provides comprehensive genomic profiling through its FoundationOne® platform, which uses ML to assess hundreds of genes for mutations and alterations relevant to targeted therapies. In a notable case involving metastatic melanoma, Foundation Medicine's profiling identified a BRAF mutation. ML algorithms recommended a targeted BRAF inhibitor, which led to substantial tumor shrinkage and extended remission for the patient.
Guardant Health specializes in liquid biopsy technology, utilizing ML algorithms to analyze circulating tumor DNA (ctDNA). This non-invasive method allows for real-time monitoring of tumor dynamics and treatment responses. A patient with lung cancer was monitored using Guardant Health’s liquid biopsy, where ML algorithms tracked ctDNA changes to adjust therapy promptly. This approach resulted in improved disease management and patient outcomes.
PathAI enhances pathology diagnoses with ML by analyzing pathology images to identify cancerous tissues and predict patient outcomes. In one application, PathAI’s algorithms assessed pathology slides from a breast cancer patient, detecting aggressive cancer subtypes that informed a more aggressive treatment plan. This precise analysis contributed to more effective management of the patient’s condition.
Precision oncology demonstrates the transformative potential of integrating machine learning with genomic data to deliver personalized cancer treatments. The examples from IBM Watson for Oncology, Tempus, Foundation Medicine, Guardant Health, and PathAI illustrate the practical applications of ML in improving cancer care through targeted therapies based on comprehensive genetic and molecular insights.
B. Pharmacogenomics
Pharmacogenomics is a field of study that examines how an individual's genetic makeup affects their response to drugs. By integrating genomic information into drug development and clinical practice, pharmacogenomics aims to enhance drug efficacy, minimize adverse drug reactions, and personalize treatment plans based on genetic profiles. The application of pharmacogenomics involves the use of machine learning (ML) and genomic data to tailor pharmacotherapy to individual genetic variations.
- Warfarin Dosing : Warfarin, an anticoagulant used to prevent blood clots, exhibits significant variability in its effectiveness and risk of bleeding among patients. Genetic variations in the VKORC1 and CYP2C19 genes influence warfarin metabolism and response. ML algorithms analyze these genetic variants along with patient data to predict the optimal warfarin dosage for each individual. For example, the Warfarin Dosing Algorithm developed by the Clinical Pharmacogenetics Implementation Consortium (CPIC) utilizes ML to integrate genetic and clinical data, helping clinicians tailor warfarin dosing more accurately. A study demonstrated that patients guided by pharmacogenomic-based dosing had fewer adverse events and more stable INR levels compared to those receiving standard dosing.
- Adverse Drug Reactions (ADRs): Pharmacogenomics can also predict the risk of adverse drug reactions (ADRs) by identifying genetic predispositions. For instance, the pharmacogenomic test for the gene HLA-B1502 is used to predict the risk of severe skin reactions to the antiepileptic drug carbamazepine. ML models analyze genetic data to identify patients who carry the HLA-B1502 allele and, consequently, are at higher risk of developing Stevens-Johnson Syndrome (SJS) or toxic epidermal necrolysis (TEN). The use of this genetic information has led to the implementation of pre-treatment screening guidelines, significantly reducing the incidence of these severe reactions.
- Antidepressant Response : The efficacy of antidepressants such as selective serotonin reuptake inhibitors (SSRIs) can vary widely among individuals. Genetic variations in the serotonin transporter gene (SLC6A4) and other related genes influence how patients respond to SSRIs. ML algorithms are used to develop pharmacogenomic tests that predict which antidepressant is likely to be most effective based on genetic profiles. For example, the GeneSight test evaluates multiple genetic markers to provide personalized recommendations for antidepressant therapy. Clinical studies have shown that patients using GeneSight-guided therapy experience faster relief from depressive symptoms compared to those receiving standard treatment.
- Cancer Chemotherapy: In oncology, pharmacogenomics helps tailor chemotherapy regimens based on genetic variations that affect drug metabolism. The enzyme thiopurine methyltransferase (TPMT) metabolizes drugs such as 6-mercaptopurine, used in leukemia treatment. Genetic variants in the TPMT gene can lead to severe toxicity or therapeutic failure. ML models analyze TPMT genetic data to guide dose adjustments, reducing the risk of adverse effects. For example, a study found that patients with low TPMT activity who received adjusted doses of 6-mercaptopurine had fewer adverse effects and improved outcomes compared to those receiving standard doses without genetic guidance.
C. Disease Risk Prediction
Disease risk prediction through the integration of machine learning (ML) and genomic data represents a significant advancement in personalized medicine. This approach involves leveraging genetic information to identify individuals at increased risk for developing specific diseases, enabling early intervention and tailored preventive strategies. By analyzing complex patterns in genetic data, ML models can provide more accurate and individualized risk assessments than traditional methods.
- Cardiovascular Disease : Artificial Intelligence can diagnose cardiovascular diseases in patients. For instance, Seah et al. utilized a neural network classifier to detect congestive heart failure from chest radiographs [60]. Cardiovascular diseases (CVDs) are influenced by both genetic and environmental factors. ML models are employed to analyze genomic data alongside lifestyle and clinical factors to predict the risk of developing CVD. For instance, the Polygenic Risk Score (PRS) is a tool that aggregates the effects of numerous genetic variants associated with CVD into a single score, which can be used to assess an individual's risk. Studies have demonstrated that individuals with high PRS scores have a significantly higher risk of CVD events compared to those with low scores. Additionally, ML algorithms can integrate data from various sources, including genome-wide association studies (GWAS) and electronic health records, to refine risk predictions and guide personalized prevention strategies.
- Breast Cancer : Genetic variants in genes such as BRCA1 and BRCA2 are well-established markers for increased breast cancer risk. ML techniques enhance risk prediction by combining genetic data with other risk factors, such as family history and lifestyle. For example, the BOADICEA model integrates genomic data with clinical and family history to estimate breast cancer risk more accurately. This model uses ML algorithms to analyze the combined effects of multiple genetic variants and interactions between them. Research has shown that incorporating ML into breast cancer risk prediction models improves the accuracy of identifying high-risk individuals, allowing for more targeted screening and preventive measures.
- Type 2 Diabetes: The prediction of type 2 diabetes risk benefits from ML algorithms that analyze genetic, lifestyle, and clinical data. Genetic variants associated with insulin resistance and beta-cell function contribute to an individual's risk of developing type 2 diabetes. ML models, such as those developed using the Diabetes Risk Score, integrate these genetic variants with other risk factors, including body mass index (BMI) and family history. These models have demonstrated improved predictive performance compared to traditional risk factors alone, enabling earlier identification of individuals at high risk and allowing for personalized lifestyle interventions to prevent disease onset.
- Alzheimer's Disease: Alzheimer's disease is influenced by both genetic predispositions and environmental factors. ML algorithms are applied to analyze genetic variants, such as those in the APOE gene, along with neuroimaging and clinical data to predict Alzheimer's disease risk. For example, the Alzheimer's Disease Neuroimaging Initiative (ADNI) utilizes ML to integrate genomic, imaging, and clinical data to develop risk prediction models. These models can identify individuals at high risk of developing Alzheimer's disease, even before clinical symptoms appear, facilitating early intervention and monitoring strategies.
- Rare Genetic Disorders: ML approaches are also useful in predicting the risk of rare genetic disorders by analyzing genomic data for specific genetic variants associated with these conditions. For instance, ML models can process data from whole exome sequencing (WES) to identify individuals who carry rare pathogenic variants linked to disorders such as cystic fibrosis or Huntington's disease. These predictive models can assist in genetic counseling and guide preventive measures for individuals and families affected by rare genetic disorders.
VI. FUTURE DIRECTIONS AND OPEN CHALLENGES
A. Emerging Trends in Machine Learning and Genomics
The integration of multi-omics data represents a significant trend in the future of machine learning (ML) applications in genomics. Multi-omics approaches involve the simultaneous analysis of various types of biological data, including genomics, transcriptomics, proteomics, and metabolomics. The goal is to achieve a more comprehensive understanding of biological systems and disease mechanisms by combining information from different molecular layers.
- Holistic Disease Understanding: Integrating data from multiple omics layers allows researchers to capture the complexity of biological systems more effectively. For instance, combining genomics (DNA sequences), transcriptomics (RNA expression profiles), and proteomics (protein levels) can provide insights into gene regulation, protein function, and metabolic pathways, offering a more detailed view of disease processes and treatment responses.
- Enhanced Predictive Models: Multi-omics integration enables the development of more robust predictive models by incorporating diverse data types. Machine learning algorithms can leverage these integrated datasets to improve the accuracy of disease risk prediction, drug response forecasting, and patient stratification. For example, integrating genomic and proteomic data can enhance the prediction of cancer progression and therapeutic outcomes.
- Data Fusion Challenges: One of the challenges in multi-omics integration is effectively fusing data from different sources with varying scales and types. Developing sophisticated algorithms that can handle heterogeneous data and extract meaningful patterns is essential for advancing multi-omics research. Addressing data integration challenges requires advancements in both computational techniques and data preprocessing methods.
- As machine learning models become increasingly complex, there is a growing emphasis on developing interpretable models that provide insights into the underlying biological processes. Interpretability is crucial for translating ML findings into actionable clinical insights and ensuring that models can be trusted and understood by researchers and clinicians.
- Explainable AI (XAI) in Genomics: Explainable AI techniques aim to make machine learning models more transparent and interpretable. For example, techniques such as feature importance analysis, partial dependence plots, and SHAP (SHapley Additive exPlanations) values help elucidate how specific features contribute to model predictions. In genomics, these methods can reveal which genetic variants or molecular features are most influential in predicting disease outcomes or treatment responses.
- Model Transparency and Validation: Ensuring the transparency and validity of ML models is essential for their adoption in clinical practice. Researchers are focusing on developing models that not only provide accurate predictions but also offer explanations that align with biological knowledge. This involves validating models against independent datasets, incorporating domain expertise, and addressing potential biases that may affect model interpretations.
- Challenges in Interpretability: Achieving interpretability in complex ML models, such as deep learning networks, remains a challenge. While deep learning models often provide high accuracy, their complexity can make it difficult to understand the rationale behind their predictions. Developing methods that balance accuracy and interpretability is an ongoing area of research.
B. Areas That Need Further Exploration and Innovation
- Integration and Standardization of Omics Data: Despite advancements, integrating multi-omics data remains a complex task due to differences in data types, formats, and scales. There is a need for standardized frameworks and methods that facilitate data integration and ensure compatibility across different omics layers. Research into standardized protocols and data-sharing platforms can enhance collaborative efforts and improve the reproducibility of findings.
- Handling Sparse and Noisy Data: Genomic data, particularly from high-throughput technologies, can be sparse and noisy. Developing robust methods for handling missing data, noise, and variability is crucial for improving the reliability of ML models. Techniques such as data imputation, noise filtering, and uncertainty quantification are areas of active research.
- Ethical and Privacy Concerns: As genomic data becomes increasingly integrated with ML models, addressing ethical and privacy concerns is paramount. Ensuring the protection of sensitive genetic information and obtaining informed consent from participants are critical issues. Research into secure data management practices, consent mechanisms, and ethical guidelines is essential for responsible data use.
C. Potential Breakthroughs on the Horizon
- Personalized Medicine Revolution: Advances in ML and genomics are poised to revolutionize personalized medicine by enabling highly individualized treatment strategies. Breakthroughs in multi-omics integration, interpretable models, and data analysis methods may lead to more precise disease diagnostics, tailored therapies, and improved patient outcomes.
- Novel Therapeutic Targets: ML-driven discoveries of novel genetic and molecular targets have the potential to drive the development of new therapies. Identifying previously unrecognized biomarkers and drug targets can lead to innovative treatments and therapeutic approaches that address unmet medical needs.
- Enhanced Patient Stratification: ML models that integrate diverse omics data may improve patient stratification, allowing for more accurate identification of patient subgroups with specific disease characteristics or treatment responses. This could lead to more effective and targeted interventions, reducing trial-and-error approaches in clinical practice.
Conclusion
The integration of machine learning (ML) in genomic medicine represents a transformative advancement in our approach to understanding and treating diseases. The advancement of precision medicine, coupled with the rise of artificial intelligence in healthcare, is steering the field towards a more individualized approach to disease management, moving away from traditional population-based methods [61]. This paper has explored the key aspects of ML in genomics, including the types of genomic data, the challenges associated with data analysis, and the critical applications in personalized medicine. In summary, ML has significantly enhanced our ability to interpret complex genomic data, providing powerful tools for predicting phenotypes, identifying genetic variants, and tailoring therapies. By leveraging various types of genomic data—such as DNA sequencing, transcriptomic profiles, and epigenomic information—researchers can build more accurate models that offer deeper insights into disease mechanisms and treatment responses. The applications of ML in personalized medicine are vast and impactful. From precision oncology, where ML aids in cancer diagnosis and drug discovery, to pharmacogenomics, which predicts drug responses based on genetic profiles, and disease risk prediction, where ML models identify genetic risk factors for various conditions. These applications underscore the potential of ML to revolutionize clinical practice by enabling more personalized and effective treatment strategies. However, challenges remain. Issues such as high dimensionality of data, noise, and integration of multi-omics datasets pose significant hurdles. Moreover, the need for interpretable ML models and addressing ethical and privacy concerns are critical areas that require ongoing research and innovation. Looking ahead, the future of ML in genomic medicine promises exciting possibilities. Advances in integrating multi-omics data, developing interpretable models, and addressing open research questions will drive the next generation of personalized medicine. By overcoming these challenges and harnessing the potential of ML, we can expect significant strides in understanding complex diseases, discovering novel therapeutic targets, and ultimately improving patient care. In conclusion, the synergy between machine learning and genomics holds the potential to unlock new dimensions in medical research and clinical practice, offering hope for more precise, effective, and personalized healthcare solutions. Continued research and collaboration in this field are essential for translating these advancements into tangible benefits for patients and the broader healthcare system.
References
[1] S. M. Metev and V. P. Veiko, Laser Assisted Microtechnology, 2nd ed., R. M. Osgood, Jr., Ed. Berlin, Germany: Springer-Verlag, 1998. [2] J. D. Watson and F. H. C. Crick, ‘‘Molecular structure of nucleic aids: A structure for deoxyribose nucleic acid,’’ Nature, vol. 171, no. 4356, pp. 737–738, 1953. [3] E. S. Lander et al., ‘‘Initial sequencing and analysis of the human genome,’’ Nature, vol. 409, no. 6822, pp. 860–921, 2001. [4] E. S. Lander, ‘‘Initial impact of the sequencing of the human genome,’’ Nature, vol. 470, no. 7333, pp. 187–197, 2011. [5] Bedi G, Carrillo F, Cecchi GA, Slezak DF, Sigman M, Mota NB, Corcoran CM. Automated analysis of free ***speech predicts psychosis onset in high-risk youths. NPJ Schizophr. 2015;1:15030. [6] Chang EK, Yu CY, Clarke R, Hackbarth A, Sanders T, Esrail- ian E, Runyon BA. Defining a patient population ***with cir- rhosis. J Clin Gastroenterol. 2016;50(10):889–94. [7] Miotto R, Li L, Kidd BA, Dudley JT. Deep patient: an unsu- pervised representation to predict the future of patients from the electronic health records. Sci Rep. 2016;6(1):1–10. [8] Osborne JD, Wyatt M, Westfall AO, Willig J, Bethard S, Gor- don G. Efficient identification of nationally mandated reporta- ble cancer cases using natural language processing and machine learning. J Am Med Inform Assoc. 2016;23(6):1077–84. [9] Garvin, J. H., Kim, Y., Gobbel, G. T., Matheny, M. E., Redd, A., Bray, B. E., & Meystre, S. M. (2018). Automating quality measures for ***heart failure using natural language process- ing: a descriptive study in the department of veterans’ affairs. JMIR medical informatics, 6(1), e9150. [10] Syrjala KL. Opportunities for improving oncology care. Lancet Oncol. 2018;19(4):449. [11] RitchieMD,deAndradeM,KuivaniemiH.Thefoundationof precision medicine: integrating electronic health records with genomics through basic, clinical, and translational research. Front Genet. 2015;6:104. [12] SbonerA,ElementoO.Aprimeronprecisionmedicineinformat- ics. Brief Bioinform. 2016;17(1):145–53. [13] Zou J, Huss M, Abid A, Mohammadi P, Torkamani A, Tel- enti A. A primer on deep learning in genomics. Nat Genet. 2019;51(1):12–8. [14] Huang S, Cai N, Pacheco PP, Narrandes S, Wang Y, Xu W. Applications of support vector machine (SVM) learning in can- cer genomics. Cancer Genomics Proteomics. 2018;15(1):41–51. [15] Cho, Gyeongcheol et al. “Review of Machine Learning Algo- rithms for Diagnosing Mental Illness.” Psychiatry investigation vol. 16,4 (2019): 262–269. doi:https://doi.org/10.30773/pi.2018. [16] Cruz JA, Wishart DS. Applications of machine learn- ing in cancer prediction and prognosis. Cancer informatics. 2006;2:117693510600200030. [17] Sampathkumar H, Chen XW, Luo B. Mining adverse drug reac- tions from online healthcare forums using hidden Markov model. BMC Med Inform Decis Mak. 2014;14(1):1–18. [18] Huang, Z., Dong, W., Wang, F., & Duan, H. (2015). Medical inpatient journey modelling and clustering: a Bayesian hidden Markov model-based approach. In AMIA Annual Symposium Proceedings (Vol. 2015, p. 649). American Medical Informatics Association. [19] Esmaili, N., Piccardi, M., Kruger, B., & Girosi, F. (2019). Cor- rection: Analysis of healthcare service utilisation after transport- related injuries by a mixture of hidden Markov models (PLoS ONE (2018) 13: 11 (e0206274. PLoS One. [20] Huang Q, Cohen D, Komarzynski S, Li XM, Innominato P, Le?vi F, Finkensta?dt B. Hidden Markov models for monitoring circa- dian rhythmicity in telemetric activity data. J R Soc Interface. 2018;15(139):20170885. [21] Marchuk Y, Magrans R, Sales B, Montanya J, Lo?pez-Aguilar J, De Haro C, Blanch L. Predicting ***patient-ventilator asynchro- nies with hidden Markov models. Sci Rep. 2018;8(1):1–7. [22] Naithani G, Kivinummi J, Virtanen T, Tammela O, Peltola MJ, Leppa?nen JM. Automatic segmentation of infant cry signals using hidden Markov models. EURASIP Journal on Audio, Speech, and Music Processing. 2018;2018(1):1–14. [23] Hasanc?ebi O, Erbatur F. Evaluation of crossover techniques in genetic algorithm based optimum structural design. Comput Struct. 2000;78(1–3):435–48. [24] Wei W, Visweswaran S, Cooper GF. The application of naive Bayes model averaging to predict Alzheimer’s disease from genome-wide data. J Am Med Inform Assoc. 2011;18(4):370–5. [25] Doing-Harris, K., Mowery, D. L., Daniels, C., Chapman, W. W., & Conway, M. (2016). Understanding patient satisfaction with received healthcare services: a natural language processing approach. In AMIA annual symposium proceedings (Vol. 2016, p. 524). American Medical Informatics Association. [26] Grover, D., Bauhoff, S., & Friedman, J. (2019). Using supervised learning to select audit targets in performance-based financing in health: An example from Zambia. PloS one, 14(1), e0211262. [27] Wagholikar, K. B., Vijayraghavan, S., & Deshpande, A. W. (2009, September). Fuzzy naive Bayesian model for medical diagnostic decision support. In 2009 Annual International Con- ference of the IEEE Engineering in Medicine and Biology Soci- ety (pp. 3409–3412). IEEE. [28] Al-Aidaroos KM, Bakar AA, Othman Z. Medical data classifica- tion with Naive Bayes approach. Inf Technol J. 2012;11(9):1166. [29] Sebastiani P, Solovieff N, Sun J. Nai?ve Bayesian classifier and genetic risk score for genetic risk prediction of a categorical trait: not so different after all! Front Genet. 2012;3:26. [30] Srinivas K, Rani BK, Govrdhan A. Applications of data mining techniques in healthcare and prediction of heart attacks. Interna- tional Journal on Computer Science and Engineering (IJCSE). 2010;2(02):250–5. [31] Altman NS. An introduction to kernel and nearest-neighbour nonparametric regression. Am Stat. 1992;46(3):175–85. [32] Zhang, Z. (2016). Introduction to machine learning: k-nearest neighbours. Annals of translational medicine, 4(11). [33] Hu LY, Huang MW, Ke SW, Tsai CF. The distance function effect on k-nearest neighbour classification for medical datasets. Springerplus. 2016;5(1):1–9. [34] Li, C., Zhang, S., Zhang, H., Pang, L., Lam, K., Hui, C., & Zhang, S. (2012). Using the K-nearest neighbour algorithm for the classification of lymph node metastasis in gastric cancer. Computational and mathematical methods in medicine, 2012. [35] Sarkar, M., & Leong, T. Y. (2000). Application of K-nearest neighbours’ algorithm on breast cancer diagnosis problem. In Proceedings of the AMIA Symposium (p. 759). American Medi- cal Informatics Association. [36] Vitola J, Pozo F, Tibaduiza DA, Anaya M. A sensor data fusion system based on k-nearest neighbour pattern classifi- cation for structural health monitoring applications. Sensors. 2017;17(2):417. [37] Zhao D, Weng C. Combining PubMed knowledge and EHR data to develop a weighted Bayesian network for pancreatic cancer prediction. J Biomed Inform. 2011;44(5):859–68. [38] Baum LE, Petrie T. Statistical inference for probabilis- tic functions of finite-state Markov chains. Ann Math Stat. 1966;37(6):1554–63. [39] Baum LE, Eagon JA. An inequality with applications to statistical estimation for probabilistic functions of Markov processes and a model for ecology. Bull Am Math Soc. 1967;73(3):360–3. [40] H. Ledford, ‘‘End of cancer-genome project prompts rethink,’’ Nature, vol. 517, no. 7533, pp. 128–129, 2015. [41] L. Cong et al., ‘‘Multiplex genome engineering using CRISPR/Cas systems,’’ Science, vol. 339, no. 6121, pp. 819–823, 2013. [42] P. Mali et al., ‘‘RNA-guided human genome engineering via Cas9,’’ Science, vol. 339, no. 6121, pp. 823–826, 2013. [43] Roth SC. What is genomic medicine? J Med Library Assoc JMLA. 2019;107(3):442. [44] Mukherjee S. The gene: an intimate history. Scribner; 2017. pp. 322–6 [45] Teng H, Cao MD, Hall MB, Duarte T, Wang S, Coin LJ. Chiron: translating raw nanopore signal directly into nucleotide sequence using deep learning. GigaScience. 2018;7(5):giy037. [46] Wick RR, Judd LM, Holt KE. Performance of neural network base calling tools for Oxford Nanopore sequencing. Genome Biol. 2019;20(1):1–10. [47] Boz?a V, Brejova? B, Vinar? T. DeepNano: deep recurrent neu- ral networks for base calling in MinION nanopore reads. PLoS ONE. 2017;12(6):e0178751. [48] K. B. Cook, T. R. Hughes, and Q. D. Morris, ‘‘High-throughput characterization of protein-RNA interactions,’’ Brief. Funct. Genomics, vol. 14, no. 1, pp. 74–89, 2014. [49] Beaulieu-Jones BK, Greene CS. Semi-supervised learning of the electronic health record for phenotype stratification. J Biomed Inform. 2016;64:168–78. [50] Basile AO, Ritchie MD. Informatics and machine learn- ing to define the phenotype. Expert Rev Mol Diagn. 2018;18(3):219–26. [51] Xu C. A review of somatic single nucleotide variant calling algorithms for next-generation sequencing data. Comput Struct Biotechnol J. 2018;16:15–24. [52] Ainscough BJ, Barnell EK, Ronning P, Campbell KM, Wag- ner AH, Fehniger TA, Griffith OL. A deep learning approach to automate refinement of somatic ***variant calling from cancer sequencing data. Nat Genet. 2018;50(12):1735–43. [53] Sahraeian SME, Liu R, Lau B, Podesta K, Mohiyuddin M, Lam HY. Deep convolutional neural networks for accurate somatic mutation detection. Nat Commun. 2019;10(1):1–10. [54] Pounraja VK, Jayakar G, Jensen M, Kelkar N, Girirajan S. A machine-learning approach for accurate detection of copy number variants from exome sequencing. Genome Res. 2019;29(7):1134–43. [55] Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat Rev Genet. 2015;16(3):172–83. [56] Li H, Siddiqui O, Zhang H, Guan Y. Cooperative learning improves protein abundance prediction in cancers. BMC Biol. 2019;17(1):1–14. [57] Coudray N, Ocampo PS, Sakellaropoulos T, Narula N, Snuderl M, Fenyo? D, Tsirigos A. Classification and mutation ***pre- diction from non–small cell lung cancer histopathology images using deep learning. Nat Med. 2018;24(10):1559–67. [58] Huttunen, M. J., Hassan, A., McCloskey, C. W., Fasih, S., Upham, J., Vanderhyden, B. C., & Murugkar, S. (2018). Auto- mated ***classification of multiphoton microscopy images of ovarian tissue using deep learning. Journal of biomedical optics, 23(6), 066002. [59] Brinker TJ, Hekler A, Enk AH, Berking C, Haferkamp S, Haus- child A, Utikal JS. Deep neural networks ***are superior to dermatologists in melanoma image classification. Eur J Cancer. 2019;119:11–7. [60] Seah JC, Tang JS, Kitchen A, Gaillard F, Dixon AF. Chest radio- graphs in congestive heart failure: visualising neural network learning. Radiology. 2019;290(2):514–22. [61] Quazi, S. (2021). An overview of CAR T cell-mediated B cell Maturation Antigen therapy.
Copyright
Copyright © 2024 Aayushi Saraswat, Shrikanth Roopesh. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET63975
Publish Date : 2024-08-14
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online