

Ijraset Journal For Research in Applied Science and Engineering Technology
NGS Analysis to Detect Mutation in Brain Tumor Diagnostic
Authors: Uma Kumari, Aastha Tanwar, Jositta George, Daityari Nayak
DOI Link: https://doi.org/10.22214/ijraset.2023.54895
Certificate: View Certificate
Abstract
This study presents an integrated computational approach for analyzing protein sequences and their 3D structures. By leveraging the MMDB Macromolecular database, homologs of a protein sequence of interest are identified, and interactive visualization of their structural properties is provided. The computational alignment method, using BLASTP, allows for efficient determination of sequence similarities and identification of conserved regions among multiple protein sequences. COBALT is employed to refine sequence alignments and facilitate graphical analysis of sequence relationships. RASMOL, a computational analysis program, generates 2-D representations of protein-ligand complexes, enabling visual exploration of their interactions. ORF finder is used to identify coding regions in mRNA sequences, aiding in the prediction of protein-coding regions. The approach is applied to brain tumor diagnostics using human biological samples, exploring the structural properties of brain tumor-related proteins with the help of the 2RHU protein structure and PYMOL visualization software. Overall, this integrated computational framework offers a comprehensive toolkit for protein sequence analysis, structure visualization, and homology modeling, with potential applications in drug discovery, molecular biology, and medical diagnostics.
Introduction
I. NTRODUCTION
Brain tumors pose unique challenges due to the intricate and delicate nature of the central nervous system. This section provides an overview of the prevalence and classification of brain tumors, emphasizing the need for comprehensive research endeavors. Next-generation sequencing (NGS) has revolutionized the field of genomics by enabling rapid and comprehensive analysis of genetic variations. Brain tumors are complex and heterogeneous diseases characterized by diverse genetic alterations. Traditional methods for mutation detection in brain tumors are often limited in scope and efficiency. NGS offers a high-throughput and comprehensive approach to analyze the entire genome, exome, or targeted gene panels, providing a more comprehensive view of genetic alterations in brain tumors. NGS analysis enables the identification of various types of genetic alterations in brain tumors, including single nucleotide variants (SNVs), insertions and deletions (indels), copy number variations (CNVs), and structural variations (SVs). These mutations can be further classified into driver mutations, which directly contribute to tumor initiation or progression, and passenger mutations, which are incidental and do not confer a selective advantage to the tumor [1,2].
While NGS analysis offers immense potential for brain tumor diagnostics, several challenges need to be addressed. These include data management and storage, bioinformatics expertise, standardization of analysis pipelines, and clinical interpretation of genetic variants. Moreover, the integration of other multi-omics data, such as transcriptomics and epigenomics, can provide a more comprehensive understanding of brain tumor biology. NGS-based mutation detection has numerous clinical applications in brain tumor diagnostics, including identifying actionable mutations, predicting therapeutic response, monitoring treatment efficacy, and detecting minimal residual disease. Furthermore, the integration of NGS analysis with emerging technologies, such as single-cell sequencing and liquid biopsy, holds promise for non-invasive tumor profiling and real-time monitoring of treatment response [3].
The three malignant brain tumor (3-MBT) repeats play a crucial role in epigenetic regulation and have been implicated in various diseases, including cancer. Epigenetic regulation plays a fundamental role in controlling gene expression patterns and maintaining cellular homeostasis. The 3-MBT repeats are key players in epigenetic regulation, with emerging evidence linking them to various diseases, particularly cancer. Understanding the structural details of the 3-MBT repeats and their interactions with relevant biomolecules is crucial for deciphering their functional roles and potential therapeutic targeting. The crystal structure of the 3-MBT repeats from human L3MBTL1 (PDB ID: 2RHU) provides a high-resolution view of the protein domain in complex with dimethyl-lysine. This structure reveals the molecular interactions between the 3-MBT repeats and dimethyl-lysine, shedding light on the recognition mechanism and specificity of this interaction [4,5]. The structural analysis unveils key residues involved in binding and highlights potential druggable sites for therapeutic interventions. The 2RHU structure also presents a unique chimera complex, where the 3-MBT repeats from L3MBTL1 are fused with histone H3.3.
This chimera structure offers valuable insights into the functional implications of the interaction between the 3-MBT repeats and histone proteins. By elucidating the structural basis of this complex, we gain a deeper understanding of the interplay between epigenetic factors and the histone code, potentially unraveling novel regulatory mechanisms in gene expression and chromatin organization. The 2RHU structure provides crucial information about the molecular interactions between the 3-MBT repeats, dimethyl-lysine, and histone H3.3. It highlights the residues involved in binding, the conformational changes induced upon complex formation, and the potential functional consequences of these interactions. These findings have implications for understanding epigenetic regulation, aberrant chromatin remodeling in diseases, and the development of targeted therapies. The crystal structure of the 3-MBT repeats from L3MBTL1 (PDB ID: 2RHU) sheds light on the structural basis of epigenetic regulation and its potential implications in disease mechanisms. Further investigations into the functional consequences of these interactions, the identification of small molecule inhibitors targeting the 3-MBT repeats, and the development of therapeutic strategies hold promise for treating diseases associated with aberrant epigenetic regulation [5,6].
The crystal structure of the 3-MBT repeats from human L3MBTL1 (PDB ID: 2RHU) in complex with dimethyl-lysine and in chimera with histone H3.3 provides valuable insights into the structural basis of epigenetic regulation. This structural information enhances our understanding of the molecular interactions and functional implications of the 3-MBT repeats, opening avenues for future research and potential therapeutic interventions in diseases involving dysregulated epigenetic mechanisms [7].
Brain cancer encompasses a diverse group of tumors arising from different cell types within the central nervous system. Understanding the underlying mechanisms and complexities of brain tumorigenesis is crucial for effective management of these malignancies. Genetic and epigenetic alterations play a pivotal role in brain tumorigenesis. We discuss key genetic mutations, such as alterations in TP53, PTEN, and IDH genes, as well as epigenetic modifications, including DNA methylation and histone modifications. These molecular changes contribute to disrupted signaling pathways, uncontrolled proliferation, and resistance to therapy. The intricate tumor microenvironment, consisting of immune cells, stromal cells, and the extracellular matrix, influences tumor progression and therapy response [8,9]. We explore the complex interactions between tumor cells and the microenvironment, highlighting the roles of immune evasion, angiogenesis, and the blood-brain barrier. Accurate diagnosis of brain cancer is crucial for appropriate treatment strategies. We discuss advanced imaging techniques, such as magnetic resonance imaging (MRI), positron emission tomography (PET), and spectroscopy, as well as molecular profiling approaches, including next-generation sequencing (NGS) and liquid biopsy, for precise characterization and classification of brain tumors. We delve into the evolving landscape of therapeutic modalities for brain cancer. This includes targeted therapies, such as inhibitors of receptor tyrosine kinases and DNA repair enzymes, immunotherapies, such as immune checkpoint inhibitors and CAR-T cell therapy, as well as emerging strategies like gene therapy and nanotechnology-based approaches. We highlight their mechanisms of action, challenges, and promising preclinical and clinical outcomes. Brain cancer remains a formidable challenge, necessitating continued research efforts to unravel its complexities. Future directions involve further elucidation of tumor heterogeneity, identification of novel therapeutic targets, and the development of personalized treatment approaches. By combining multidisciplinary efforts and leveraging technological advancements, we strive for improved outcomes and enhanced quality of life for patients affected by brain cancer [9,10].
Next-generation sequencing (NGS) has revolutionized the field of genomics, enabling rapid, cost-effective, and high-throughput analysis of DNA and RNA. Next-generation sequencing (NGS) platforms have surpassed traditional sequencing methods, allowing comprehensive analysis of the genome, transcriptome, and epigenome. We introduce the fundamental principles of NGS technology and its impact on genomics research and healthcare. The evolution of NGS has led to increased sequencing capacity, reduced costs, and improved data quality, making it more accessible to researchers and clinicians. NGS has transformed human genetics research, enabling the identification of disease-causing variants, genome-wide association studies (GWAS), and rare variant discovery [11,12]. We explore how NGS has revolutionized Mendelian disorders, complex diseases, and population genetics studies, enhancing our understanding of the genetic basis of human traits and diseases. NGS has propelled cancer genomics, facilitating the identification of somatic mutations, driver genes, and genomic alterations associated with tumorigenesis. We discuss how NGS-based approaches, including whole-exome sequencing (WES), whole-genome sequencing (WGS), and targeted gene panels, have contributed to precision oncology, prognostication, and therapeutic decision-making [13].
The nucleosome, consisting of DNA wrapped around a histone octamer, is the fundamental unit of chromatin. The histone proteins H2A, H2B, H3, and H4 form the core of the nucleosome and undergo various post-translational modifications, such as acetylation, methylation, phosphorylation, and ubiquitylation. These modifications on the histone tails play critical roles in regulating transcription and maintaining genome integrity. Among the proteins involved in chromatin regulation, the MBT (Malignant Brain Tumor) repeat is a conserved structural motif found in various organisms, including humans. There are at least 9 MBT repeat proteins in the human genome, and they exist as tandem repeats within these proteins.
The discovery of the MBT repeat came from studying the Drosophila tumor-suppressor protein L(3)MBT, which, when mutated, leads to malignant transformations of optic neuroblasts. MBT repeat proteins are known to recognize methylated lysine residues on histones, similar to other members of the "Royal Family" of chromatin-binding proteins. These proteins have been implicated in the regulation of chromatin transcriptional states. Structural studies have shown that MBT repeat proteins utilize a semi-aromatic cage to accommodate the methylated lysine, distinguishing between different lysine methylation states [14,15]. Understanding the structural basis of MBT repeat proteins' interactions with methylated lysine residues provides insights into their roles in chromatin regulation and gene expression. Further investigations into the functional implications of these interactions and their impact on chromatin dynamics will deepen our understanding of epigenetic regulation and its involvement in various biological processes [16, 17].
The 3-MBT (three malignant brain tumor) repeats are a conserved structural motif involved in epigenetic regulation, particularly in recognizing methylated lysine residues on histones. The 3-MBT repeats are a conserved motif implicated in the recognition of methylated lysine residues on histones. These repeats play a critical role in epigenetic regulation and have been associated with various diseases, including brain tumors. Understanding the structural details of the 3-MBT repeats and their interactions with histones is crucial for deciphering their functional roles and potential therapeutic targeting. The crystal structure of the 3-MBT repeats from human L3MBTL1 (PDB ID: 2RHU) reveals the intricate molecular interactions between the repeats and dimethyl-lysine. This structure provides a detailed view of the binding pocket and the residues involved in recognizing and accommodating the dimethylated lysine residue. The structural analysis sheds light on the specificity and affinity of the 3-MBT repeats for methylated histones. The 2RHU structure also presents a chimera complex where the 3-MBT repeats from L3MBTL1 are fused with histone H3.3. This chimera structure offers valuable insights into the functional implications of the interaction between the 3-MBT repeats and histone proteins. By elucidating the structural basis of this complex, we gain a deeper understanding of the interplay between epigenetic factors and the histone code, potentially unraveling novel regulatory mechanisms in gene expression and chromatin organization. The crystal structure of 2RHU provides crucial information about the molecular interactions between the 3-MBT repeats, dimethyl-lysine, and histone H3.3. It reveals the key residues involved in binding, the conformational changes induced upon complex formation, and the specificity of the 3-MBT repeats for methylated histones. These structural insights contribute to our understanding of the epigenetic regulatory mechanisms mediated by the 3-MBT repeats. The structural elucidation of 2RHU enhances our understanding of the functional implications of the 3-MBT repeats in epigenetic regulation. The binding of the 3-MBT repeats to methylated histones is thought to act as a marker that recruits proteins to specific regions of chromatin. By deciphering the structural details, we gain valuable insights into the mechanisms underlying epigenetic regulation and its potential implications in gene expression and genome integrity [5,6,7].
II. MATERIALS AND METHODS
The protein structure has been downloaded using PDB (protein Databank). The Protein Data Bank (PDB) stands as the central repository of experimentally determined three-dimensional structures of proteins, nucleic acids, and other biological macromolecules [18]. PDB id 2RHU is converted into amino acid sequence in FASTA format using MMDB. The Molecular Modeling Database, commonly known as MMDB, is a comprehensive repository of experimentally determined three-dimensional structures of proteins, nucleic acids, and complex assemblies. Developed and maintained by the National Center for Biotechnology Information (NCBI), MMDB integrates data from several sources, including the Protein Data Bank (PDB), which serves as the primary archive for macromolecular structure data [19]. BLASTp, an extension of the widely used BLAST algorithm, is specifically designed to compare a protein query sequence against a vast database of protein sequences. Developed by the National Center for Biotechnology Information (NCBI), BLASTp provides researchers with a powerful tool for sequence similarity searches and functional annotation [20]. ORF-Finder is a computational tool designed to identify and predict the location of potential ORFs within DNA or RNA sequences. Developed to assist in gene discovery and genome annotation, ORF-Finder plays a crucial role in deciphering the functional components of genomes [21]. SMARTBLAST leverages contextual information to provide a more refined and meaningful analysis of sequence similarities. It takes into account biological context, such as gene annotations, protein domains, and evolutionary relationships, to uncover deeper insights into the functional implications of sequence matches [20]. PyMOL is a versatile and user-friendly molecular visualization software that provides a wealth of tools and features for studying macromolecular structures. Developed by Schrödinger, PyMOL is widely used by scientists across various disciplines, including biochemistry, drug discovery, and structural biology [22,23]. RasMol, short for "Ras macromolecular visualization," is an open-source software package that allows researchers to visualize and analyze biomolecular structures.
Developed in the early 1990s by Roger Sayle, RasMol has become a widely adopted tool, offering insights into the molecular world with its user-friendly interface and powerful visualization capabilities [24,25]. The Catalogue of Somatic Mutations in Cancer (COSMIC) serves as an indispensable resource for researchers investigating the genetic alterations underlying cancer development and progression. Developed and maintained by the Wellcome Trust Sanger Institute, COSMIC provides a comprehensive and curated repository of somatic mutations identified in various cancer types. COSMIC contains a vast collection of data derived from a multitude of sources, including high-throughput sequencing projects, research publications, and public databases. The data is meticulously curated, ensuring its accuracy and reliability. With over millions of mutation records, COSMIC encompasses a broad spectrum of cancer types, providing researchers with a comprehensive view of the genomic landscape of cancer [26].
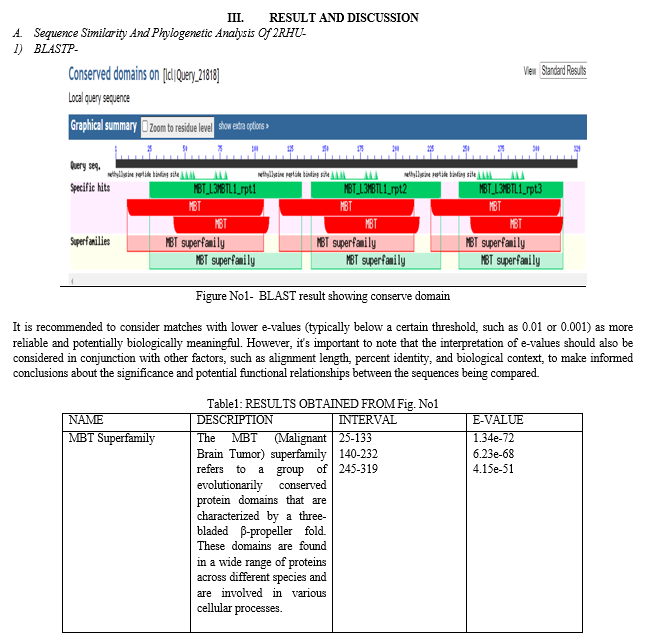


Conclusion
In conclusion, this study presents an innovative and integrated computational approach for analyzing protein sequences and their corresponding 3D structures. By utilizing the extensive MMDB Macromolecular database, the approach efficiently identifies homologs of a target protein sequence and provides interactive visualization of their structural properties. Through the use of BLASTP and COBALT, sequence alignments and conserved regions among multiple protein sequences can be accurately determined. The application of RASMOL enables the generation of 2-D representations of protein-ligand complexes, facilitating visual exploration of their interactions. Additionally, ORF finder aids in the prediction of protein-coding regions in mRNA sequences. The study demonstrates the practical application of this computational framework in brain tumor diagnostics, utilizing human biological samples. By leveraging the 2RHU protein structure and PYMOL visualization software, the structural properties of brain tumor-related proteins are explored. Overall, this integrated computational toolkit offers a comprehensive solution for protein sequence analysis, structure visualization, and homology modeling. Its potential applications extend to various fields such as drug discovery, molecular biology, and medical diagnostics. By providing valuable insights into protein structure-function relationships, this approach can contribute to advancing our understanding of complex biological systems and support the development of new therapeutic strategies.
References
[1] Sahm, F., Schrimpf, D., Jones, D.T.W. et al. Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. ActaNeuropathol 131, 903–910 (2016). https://doi.org/10.1007/s00401-015-1519-8 [2] Hendrikus J. Dubbink and others, Molecular classification of anaplastic oligodendroglioma using next-generation sequencing: a report of the prospective randomized EORTC Brain Tumor Group 26951 phase III trial, Neuro-Oncology, Volume 18, Issue 3, June 2015, Pages 388–400, https://doi.org/10.1093/neuonc/nov182 [3] Alexandra M Miller and others, Next-generation sequencing of cerebrospinal fluid for clinical molecular diagnostics in pediatric, adolescent and young adult brain tumor patients, Neuro-Oncology, Volume 24, Issue 10, October 2022, Pages 1763–1772, https://doi.org/10.1093/neuonc/noac035 [4] Li, H., Fischle, W., Wang, W., Duncan, E. M., Liang, L., Murakami-Ishibe, S., Allis, C. D., & Patel, D. J. (2007). Structural basis for lower lysine methylation state-specific readout by MBT repeats of L3MBTL1 and an engineered PHD finger. Molecular cell, 28(4), 677–691. https://doi.org/10.1016/j.molcel.2007.10.023 [5] Goedegebuure, P. S., Douville, L. M., Li, H., Richmond, G. C., Schoof, D. D., Scavone, M., &Eberlein, T. J. (1995). Adoptive immunotherapy with tumor-infiltrating lymphocytes and interleukin-2 in patients with metastatic malignant melanoma and renal cell carcinoma: a pilot study. Journal of Clinical Oncology, 13(8), 1939-1949. [6] Lenzhofer, R., Micksche, M., Dittrich, C., GRANINGER, W., JAKESZ, R., & Kolb, R. (1984). Recombinant human in IFN alpha 2 (RHU-IFN-ALPHA 2) in advanced breast cancer. Drugs under experimental and clinical research, 10(7), 463-470. [7] Grauffel, C., Stote, R. H., &Dejaegere, A. (2010). Force field parameters for the simulation of modified histone tails. Journal of computational chemistry, 31(13), 2434-2451. [8] Koo, Y. E. L., Reddy, G. R., Bhojani, M., Schneider, R., Philbert, M. A., Rehemtulla, A., ...&Kopelman, R. (2006). Brain cancer diagnosis and therapy with nanoplatforms. Advanced drug delivery reviews, 58(14), 1556-1577. [9] delBurgo, L. S., Hernández, R. M., Orive, G., &Pedraz, J. L. (2014). Nanotherapeutic approaches for brain cancer management. Nanomedicine: Nanotechnology, Biology and Medicine, 10(5), e905-e919. [10] Taphoorn, M. J., Claassens, L., Aaronson, N. K., Coens, C., Mauer, M., Osoba, D., ...& EORTC Quality of Life Group. (2010). An international validation study of the EORTC brain cancer module (EORTC QLQ-BN20) for assessing health-related quality of life and symptoms in brain cancer patients. European Journal of Cancer, 46(6), 1033-1040. [11] Tan, B., Ng, C., Nshimyimana, J. P., Loh, L. L., Gin, K. Y. H., & Thompson, J. R. (2015). Next-generation sequencing (NGS) for assessment of microbial water quality: current progress, challenges, and future opportunities. Frontiers in microbiology, 6, 1027. [12] Vincent, A. T., Derome, N., Boyle, B., Culley, A. I., &Charette, S. J. (2017). Next-generation sequencing (NGS) in the microbiological world: How to make the most of your money. Journal of microbiological methods, 138, 60-71. [13] Patel, R. K., & Jain, M. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PloS one, 7(2), e30619. [14] Eryilmaz, J., Pan, P., Amaya, M. F., Allali-Hassani, A., Dong, A., Adams-Cioaba, M. A., Mackenzie, F., Vedadi, M., & Min, J. (2009). Structural studies of a four-MBT repeat protein MBTD1. PloS one, 4(10), e7274. https://doi.org/10.1371/journal.pone.0007274 [15] Jenuwein, T., & Allis, C. D. (2001). Translating the histone code. Science (New York, N.Y.), 293(5532), 1074–1080. https://doi.org/10.1126/science.1063127 [16] Ruthenburg, A. J., Li, H., Patel, D. J., & Allis, C. D. (2007). Multivalent engagement of chromatin modifications by linked binding modules. Nature reviews. Molecular cell biology, 8(12), 983–994. https://doi.org/10.1038/nrm2298 [17] Bracken, A. P., Dietrich, N., Pasini, D., Hansen, K. H., &Helin, K. (2006). Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes & development, 20(9), 1123–1136. https://doi.org/10.1101/gad.381706 [18] Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N., & Bourne, P. E. (2000). The Protein Data Bank. Nucleic acids research, 28(1), 235–242. https://doi.org/10.1093/nar/28.1.235 [19] Thomas Madej and others, MMDB: 3D structures and macromolecular interactions, Nucleic Acids Research, Volume 40, Issue D1, 1 January 2012, Pages D461–D464, https://doi.org/10.1093/nar/gkr1162 [20] McGinnis, S., & Madden, T. L. (2004). BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic acids research, 32(Web Server issue), W20–W25. https://doi.org/10.1093/nar/gkh435 [21] Rombel, I. T., Sykes, K. F., Rayner, S., & Johnston, S. A. (2002). ORF-FINDER: a vector for high-throughput gene identification. Gene, 282(1-2), 33–41. https://doi.org/10.1016/s0378-1119(01)00819-8 [22] Seeliger, D., & de Groot, B. L. (2010). Ligand docking and binding site analysis with PyMOL and Autodock/Vina. Journal of computer-aided molecular design, 24(5), 417–422. https://doi.org/10.1007/s10822-010-9352-6 [23] Dr Uma kumari, Devanshi Gupta, In silico RNA aptamer drug design and modelling,2022/4, Journal-JETIR,Volume-9, Issue-4, Pages 718-725 [24] Roger A. Sayle,E.James Milner-White,RASMOL: biomolecular graphics for all,Trends in Biochemical Sciences,Volume 20, Issue 9,1995,Pages 374-376,ISSN 0968-0004, https://doi.org/10.1016/S0968-0004(00)89080-5 [25] Uma kumara, Navjot KaurVirk, Identification of new potential drug for lung adenocarcinoma causing protein RMB10 using computer aided drug design approach, Publication date- 2022/6/11, Journal-IJBTR, Volume-12, Issue-2, Pages 1-8 [26] Forbes, S. A., Beare, D., Gunasekaran, P., Leung, K., Bindal, N., Boutselakis, H., Ding, M., Bamford, S., Cole, C., Ward, S., Kok, C. Y., Jia, M., De, T., Teague, J. W., Stratton, M. R., McDermott, U., & Campbell, P. J. (2015). COSMIC: exploring the world\'s knowledge of somatic mutations in human cancer. Nucleic acids research, 43(Database issue), D805–D811. https://doi.org/10.1093/nar/gku1075
Copyright
Copyright © 2023 Uma Kumari, Aastha Tanwar, Jositta George, Daityari Nayak. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET54895
Publish Date : 2023-07-21
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online