

Ijraset Journal For Research in Applied Science and Engineering Technology
Advancements in Pharmacotherapy: Novel Approaches for Treating Sickle Cell Disease
Authors: Nancy ., Harmanjot Kaur, Dr. Shivam Choudghal, Pardeep Kumar
DOI Link: https://doi.org/10.22214/ijraset.2024.64436
Certificate: View Certificate
Abstract
A genetic condition known as sickle cell disease (SCD) is marked by the abnormal production of haemoglobin, which results in the formation of sickle-shaped red blood cells. This can lead to vaso-occlusive events, causing a variety of clinical complications. While traditional management strategies have focused on symptom relief and supportive care, recent years have witnessed remarkable advancements in pharmacotherapeutic approaches targeting the underlying genetic and molecular mechanisms of SCD. The aim of this review article is to provide an in-depth analysis of the emerges trends and development in pharmacotherapeutic strategies for SCD, including gene therapy, gene editing, fetal hemoglobin induction, nitric oxide pathway modulation, adhesion molecule inhibition, and novel anti-sickling agents. It reviews innovative pharmacotherapeutic interventions, including gene editing technologies, novel drug targets, and advanced therapies, emphasizing their potential in alter the course of the illness, minimize complications, and enhance the quality lifespan for individuals with sickle cell disease. We also discuss the challenges, limitations, and future directions in the development and implementation of these strategies are addressed. Background: Sickle cell disease is a genetic hemoglobinopathy characterized by the production of abnormal hemoglobin S (HbS), which causes red blood cells to deform into a sickle shape. These sickled cells are prone to hemolysis, causing anemia, and they obstruct small blood vessels, leading to vaso-occlusive crises and resultant pain, organ damage, and increased mortality. The disease predominantly affects individuals of African, Mediterranean, Middle Eastern, and Indian ancestry, with millions of people worldwide suffering from its debilitating effects. The pathophysiology of SCD is intricate and involves a series of events initiated by the polymerization of deoxygenated HbS. This polymerization causes red blood cells to deform, adhere to the vascular endothelium, and subsequently trigger inflammation, oxidative stress, and activation of blood clotting pathway. These processes collectively contribute to the acute and chronic complications seen in SCD patients, including acute pain episodes, chronic pain, stroke, acute chest syndrome, and multi-organ damage. Historically, treatment options for SCD have been limited and focused primarily on symptom management and the prevention of complications. The mainstays of therapy have included blood transfusions, hydroxyurea, and, more recently, L-glutamine. Blood transfusions help decrease the proportion of sickled cells but come with risks such as iron overload and all immunization. Hydroxyurea, a chemotherapeutic agent, stimulate the production of fetal hemoglobin (HbF), which reduce HbS polymerization. However, its use is limited by side effects and variable patient response. In 2017, FDA approved L-glutamine which helps reduce oxidative stress but does not address all the underlying pathophysiological mechanisms of SCD. In recent years, significant advancements in our understanding of the molecular and cellular mechanisms underlying SCD have paved the way for novel therapeutic approaches. These advancements include gene therapy, novel pharmacologic agents targeting specific pathways involved in the disease process, and approaches aimed at modifying the genetic defect itself. The advent of these innovative therapies holds the promise of transforming SCD from a previously life-threatening illness to a now manageable chronic disease. This article aims to explore recent advances in pharmacotherapy for SCD, focusing on novel approaches that offer hope for more effective and comprehensive management of this challenging disorder. By examining the mechanisms, efficacy, and safety of these emerging treatments, we seek to highlight their potential to improve the standard of life and outcomes for those affected by SCD.
Introduction
I. INTRODUCTION
Millions of individual worldwide are impacted by sickle cell disease, which is still a serious global health concern. The genetic mutation causing SCD results in the aberrant production of hemoglobin, resulting to a cascade of events that end in vaso-occlusive crises and multi-organ damage.1 Historically, management has focused on palliative measures to alleviate symptoms and complications. However, recent breakthroughs in biotechnology and molecular medicine have paved the way for innovative pharmacotherapeutic interventions that target the root causes of sickle cell illness.3 The characteristic feature of SCD is the transformation of usually malleable red blood cells into rigid, sickle-shaped cells, triggering vaso-occlusive crises, agonizing pain, and progressive organ damage. The burden of SCD extends globally, with millions affected and diverse clinical manifestations ranging from mild to severe.10
The relentless nature of SCD has fueled a fervent pursuit of more effective therapeutic interventions. The advent of the evolving pharmacotherapeutic landscape has rekindled optimism within the medical community and among patients alike. Recognizing the inadequacies of traditional management approaches, researchers have embarked on a journey to target the underlying molecular intricacies of SCD.2 This review aims to navigate this journey, delving into the burgeoning realm of treatment strategies that hold the promise of transforming the outlook for SCD patients.
The subsequent parts explain delve into the historical context of SCD treatment, dissect the mechanisms and potentials of novel therapies, and delve into the multifaceted challenges and opportunities that lie ahead.11 The Current analysis of the developing pharmacotherapeutic landscape, this review aim to offer an understanding of the monumental shifts underway in the pharmacotherapeutics of SCD.4
A. Traditional Approaches and Limitations
The drug hydroxyurea is a medication that has been primarily used in the treatment of hematological disorders, most notably sickle cell disease and several forms of cancer.5 It exerts its therapeutic agents by as we approach the dawn of a new era in sickle cell disease management, our investigation not only clarifies state of the interventions but also emphasizes the critical importance of adapting and advancing treatment modalities.8 Its effect on DNA synthesis and cell division, which leading to the inhibition of cell proliferation. The clinical outcomes and mechanism of action of hydroxyurea in sickle cell disease are as follows:
B. Mechanism of Action
Hydroxyurea inhibiting the enzyme ribonucleotide reductase, which is involved in the conversion of ribonucleotides to deoxyribonucleotides. Because of this inhibition, fewer reduces the availability of deoxyribonucleotides, which are necessary for DNA synthesis and repair.15 Cell division is disrupted, leading to reduced production of red blood cells with abnormal shapes (sickle cells) in sickle cell disease.
One type of haemoglobin that is specific to foetal tissue is called foetal haemoglobin (HbF) that normally present in newborns but diminishes as a person grows older. The molecular makeup of fetal hemoglobin differs from that adult hemoglobin and is less likely to forming the characteristic rigid (hard) & sickle-shaped cells.14
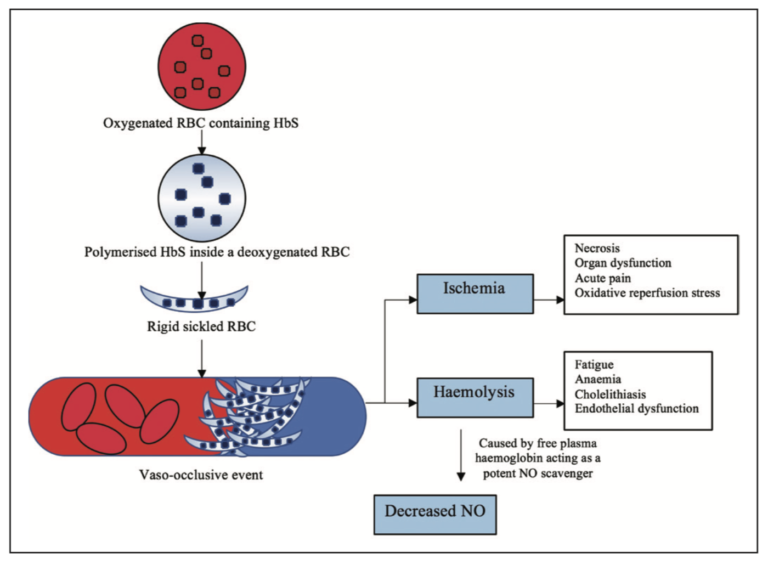
Fig: 1 Pathogenesis of sickle cell disease
II. CLINICAL OUTCOMES
Hydroxyurea has been linked with several beneficial clinical outcomes, such as:
- Reduced Pain Crises: Hydroxyurea has been demonstrated to lessen the occurrence and intensity of painful vaso-occlusive incident, a characteristic feature of sickle cell disease.20 These crises occur due to the blockage of blood vessels by sickle-shaped red blood cells, leading to organ damage and discomfort.
- Improved Anemia: Hydroxyurea can alleviate the inhance in hemoglobin levels and a reduce in reticulocyte count (immature red blood cells), addressing the anemia commonly seen in sickle cell disease.2
- Decreased Incidence of Acute Chest Syndrome: Acute chest syndrome, a severe complication of sickle cell disease marked by chest discomfort, fever, and respiratory issues, has been linked to a lower likelihood in patients undergoing hydroxyurea treatment.7
- Prevention of Stroke: Among pediatric individuals diagnosed with sickle cell disease, hydroxyurea has shown promise in reducing the risk of stroke.
A. Challenges and Limitations of Hydroxyurea Therapy
Despite its benefits, hydroxyurea therapy also comes with several challenges and limitations:
- Variable Response: The response to hydroxyurea varies among patients with sickle cell disease. The level of increase in HbF and the clinical benefits can vary from person to person.16
- Long-Term Safety: The long-term safety of hydroxyurea use is still a topic of ongoing research.11 Concerns have been raised about potential risks, include bone marrow suppression and heightened susceptibility to specific cancers.
- Adherence: Hydroxyurea is a daily oral medication, and adherence to the treatment regimen can be challenging for some patients, especially considering the potential for side effects.13
- Monitoring: Regular monitoring is required to adjust the dosage and assess the treatment's effectiveness and safety. This can be burdensome for patients and healthcare providers.
- Pregnancy and Fertility: The use of hydroxyurea during pregnancy raises concerns about potential risks to both the mother and the developing fetus. It can also affect fertility in both men and women.21
- Limited Availability: In some regions, access to hydroxyurea might be limited, preventing patients from benefiting from this treatment.
Table: -1
A clear distinction between the difficulties and constraints of employing hydroxyurea treatment for sickle cell disease.
|
S.No |
Challenges of Hydroxyurea |
Limitations of Hydroxyurea Therapy |
|
1. |
Patient Adherence: Requires long-term commitment and consistent use. Daily medication can be challenging. |
Individual Response: Effectiveness varies among patients. Some patients may not respond well. |
|
2. |
Side Effects: Typical adverse effects encompass nausea, fatigue, and myelosuppression, while a subset of patients may encounter more severe reactions. |
Cytotoxicity: Hydroxyurea is a cytotoxic drug, potentially affecting healthy cells along with sickle cells. |
|
3. |
Dose Adjustment: Finding the right dosage requires careful monitoring and adjustment. Underdosing may be ineffective, while overdosing can lead to toxicity. |
Renal Impairment: Hydroxyurea is excreted by the kidneys, and caution is needed in patients with renal impairment. |
|
4. |
Fertility Concerns: Potential impact on fertility, which can be a concern for some patients. Discussing family planning is crucial. |
Risk of Secondary Malignancies: Long-term usage could potentially elevate the risk of developing secondary malignancies. |
III. DISEASE-MODIFYING THERAPIES:
These therapies aim to address various aspects of the disease and improve the well-being of individuals with sickle cell disease and elevate their quality of life. Let's briefly discuss each of these therapies:
- Voxelotor: Voxelotor is a medication that works by increasing the levels of fetal hemoglobin (HbF) in individuals with sickle cell disease. HbF is a type of hemoglobin that is normally present in newborns and aids in preventing the formation of red blood cells from becoming misshapen and causing the characteristic sickling seen in SCD.8 By increasing HbF levels, voxelotor helps to reducing the formation of sickled red blood cells and the associated complications.
- Crizanlizumab: Crizanlizumab is a monoclonal antibody that targets adhesion molecules involved in the vaso-occlusive process in sickle cell disease. Vaso-occlusion happens when sickled red blood cells aggregate and obstruct blood vessels, resulting in pain and organ damage.22 By inhibiting these adhesion molecules, crizanlizumab strives to lower the occurrence of vaso-occlusive crises and associated severe pain.
- Luspatercept: Luspatercept is a medication primarily used to manage anemia in patients with certain types of blood disorders, including SCD.10 Anemia is a frequent side effect of SCD that can cause to fatigue and other health problems. Luspatercept works by promoting red blood cell production, which can help alleviate anemia and improve overall well-being in individuals with SCD.9
A. Emerging Frontiers: Gene Therapy and Editing
In recent years, gene therapy and editing have become prominent as promising frontiers in the field of medical science, contribution to the potential treatment for a broad spectrum of genetic disorder. Two prominent approaches in this field are lentiviral-based gene therapy and CRISPR-Cas9 technology.25 These methods hold the potential to correct genetic mutations and alleviate the underlying causes of various diseases, particularly those caused by single-gene defects.
B. Lentiviral-Based GENE Therapy
Lentiviral-based gene therapy involves using modified lentiviruses to deliver functional copies of genes into the patient's cells. This allows the corrected gene to be permanently integrated into the patient's cells, theoretically providing a lasting therapeutic effect.22
This approach has shown success in correcting genetic mutations responsible for diseases like severe combined immunodeficiency (SCID) and certain forms of inherited blindness.
By introducing functional copies of the mutated gene into the patient's cells, researchers aim to restore the normal cellular functions and alleviate disease symptoms.
However, challenges remain, such as ensuring precise targeting of the correct cells, avoiding unintended insertions of the corrected gene, and managing potential immune responses triggered by the viral vectors.
C. Crispr-Cas9 Technology
CRISPR-Cas9 technology has been transformed the field of genetic editing. It is an accurate and flexible instrument that enable researchers to precisely target specific genes and modify particular gene within the DNA of an organism and make precise modifications. Regarding genetic illnesses, CRISPR-Cas9 can be used to edit the DNA sequence, either by correcting a mutation, introducing a functional gene, or disrupting a harmful gene.16
For instance, in the case of Beta-thalassemia, a genetic condition marked by impaired β-globin production, CRISPR-Cas9 can be employed to precisely edit the β-globin gene, restoring normal hemoglobin production and potentially curing the disease.
D. Promising Results and Challenges
Both lentiviral-based gene therapy and CRISPR-Case technology has demonstrated promising results in preclinical and clinical studies. Successful treatments have been reported for some genetic disorders, offering hope for patients and their families.17
However, challenges persist. Off-target effects, where genetic editing occurs at unintended locations, remain a concern with CRISPR-Cas9. Ensuring the safety and efficacy of these therapies in the long term is a critical aspect of their development. Additionally, ethical considerations surround the genetic modification of human embryos and germline editing, requiring careful ethical deliberation and oversight.
IV. MANAGING PAIN AND COMPLICATIONS
Pain Management Strategies for Vaso-Occlusive Crises: Vaso-occlusive emergencies are one of the trademark characteristic of sickle cell illness and can cause intense pain. These episodes happen when blood vessels are obstructed by sickle-shaped red blood cells, resulting in diminished blood circulation and harm to tissues.23 Effective pain control is essential for enhancing of life for people with SCD during these crises. Several approaches include:
- Hydration: Staying well-hydrated helps prevent the clumping of sickle cells and can decrease the frequency and severity of vaso-occlusive episodes.
- Pain Medications: Pain relievers are commonly used to manage pain during crises. Non-opioid analgesics such as nonsteroidal anti-inflammatory drugs (NSAIDs) like ibuprofen suitable for mild to moderate management. Opioid medications are typically reserved for severe pain and advisable to utilize them with the supervision of a medical expert. 19
- Hydroxyurea: This medication promotes the synthesis of fetal hemoglobin, thus inhibiting the sickling of red blood cells. It can decrease the frequency and severity of vaso-occlusive episodes.
- Blood Transfusions: In extreme circumstances, blood transfusions may be necessary to improve oxygen delivery to tissues and decrease the number of sickle cells.
- Supplemental Oxygen: Administering oxygen can help improve tissue oxygenation and reduce pain during crises.
A. Non-Opioid Analgesics and Alternative Approaches:
Given the concerns about opioid addiction and side effects, non-opioid analgesics and alternative approaches are being explored to manage pain in SCD patients:
- Non-NSAID Pain Relievers: Acetaminophen (paracetamol) is a substitute for NSAIDs in the treatment of agony in SCD patients. It's crucial to remember that NSAIDs can potentially worsen kidney function in some SCD patients.8
- Nerve Blocks: Local anesthetic nerve blocks can offer specific pain relief for specific painful areas.
- Physical Therapy: Techniques such as massage, stretching, and exercise can help manage reduce discomfort and increase mobility.
- Cognitive Behavioral Therapy (CBT): CBT can assist individuals in managing their pain through relaxation techniques, mental imagery, and mindfulness.11
- Mind-Body Practices: Practices like yoga and meditation can help individuals manage pain by promoting relaxation and reducing stress.
B. Addressing Long-Term Complications of SCD:
Sickle cell disease can result in a variety of long-term difficulty that necessitate continuous management:
- Stroke Prevention: Individuals with SCD are at a greatest risk of stroke. Regular blood transfusions and the use of medications like hydroxyurea can help reduce this risk.
- Organ Damage: SCD can cause harm to a number of various organs, including the kidneys, lungs, and liver. Regular medical check-ups are essential to monitor and manage these complications.17
- Infection Prevention: SCD patients are more susceptible to infections. Vaccinations, prophylactic antibiotics, and prompt treatment of infections are important preventive measures.
- Regular Monitoring: Routine blood tests and imaging are crucial for monitoring organ function and detecting potential complications early.
- Pulmonary Hypertension Management: Pulmonary hypertension is a serious complication in some individuals with SCD. Medications and lifestyle changes may be prescribed to manage this condition.19
V. PATIENT-CENTERED CARE AND MULTIDISCIPLINARY APPROACHES
Patient-centered care has gained significant importance in healthcare, emphasizing the need to focus on the individual's preferences, needs, and values when making decisions about their health. This approach is particularly crucial in the context of individuals with the complex genetic disease known as sickle cell disease that can have a wide range of physical, emotional, and social impacts.
Holistic Care for SCD Patients: Shifting towards holistic care means examining the patient as a whole, rather than just focusing on their medical illness. For SCD patients, this involves recognizing that their well-being is influenced by various factors beyond their physical symptoms.24
This includes their emotional state, social support system, lifestyle, cultural background, and more. Holistic care for SCD patients involves not only treating the physical symptoms of the disease but also addressing their psychological and emotional needs, as well as their overall quality of life.1
Comprehensive Care Teams: Managing SCD effectively requires a multidisciplinary approach involving a team of healthcare professionals with diverse expertise. This team could include hematologists, nurses, pain specialists, social workers, psychologists, genetic counselors, nutritionists, and others.16
Mental Health Support: A patient’s mental health may suffer as a result of having a chronic illness such as SCD. Chronic pain, frequent hospital visits, and the uncertainty of the disease's progression can cause to anxiety, depression, and other psychological issues. Integrating mental health support into the care plan is essential. Mental health professionals can help patients and their families cope with the emotional and psychological impact of the disease, develop strategies to manage stress and anxiety, and improve overall mental well-being.20
Patient Education and Empowerment: Empowering SCD patients with knowledge about their condition is crucial. When patients understand their disease and treatment options, they can actively engage in making decision about their care. Education helps patients manage their symptoms, adhere to treatment plans, and recognize when they need medical attention.15 Additionally, involving patients in their care can foster a sense of control and enhance their encounter with the healthcare system as a whole.
Cultural Sensitivity: SCD affects individuals from various cultural backgrounds, and cultural factors can affect how individuals view and treat their illness. Healthcare professionals should be aware of cultural difference, practices, and values can impact treatment decisions and communication. Incorporating cultural competence into care plans can improve patient trust, adherence, and outcomes.5
VI. CHALLENGES AND FUTURE DIRECTIONS
- Accessibility and Affordability of Novel Therapies: Developing new and innovative therapies often involves significant research and development costs. As these therapies become available, ensuring they are accessible and affordable for a wide range of patients is a challenge. Regulatory agencies, governments, and healthcare organizations need to work together to balance the need for fair pricing with the financial sustainability of healthcare systems.
- Long-term Safety and Monitoring Considerations: As new therapies are introduced, it's crucial to monitor their long-term safety and effectiveness. Some therapies might have unforeseen side effects or interactions that only become apparent after extended use. Robust post-market surveillance systems and real-world data collection are essential to identify and address any safety concerns that may arise.
- Individualized Treatment Approaches Based on Genetic Factors: The field of personalized medicine is rapidly advancing, and genetic factors play a significant role in how individuals respond to treatments. Tailoring therapies to a patient's genetic makeup can enhance their efficacy and minimize adverse effects. However, implementing individualized treatment approaches on a large scale requires advancements in genetic testing technologies, data analysis, and regulatory frameworks to ensure privacy and ethical considerations.
- Collaborative Research and Patient Advocacy: Driving Advancements in Healthcare In recent years, the landscape of medical research and healthcare has evolved significantly, placing a stronger emphasis on collaboration between researchers, clinicians, and patients. This collaborative approach has proven to be instrumental in driving advancements, improving patient outcomes, and accelerating the development of innovative treatments and therapies.
A. Importance of Collaboration
Cross-disciplinary Insights: Collaboration brings together experts from various fields, such as biomedical research, clinical medicine, engineering, and social sciences. This multidisciplinary approach fosters the exchange of diverse perspectives and expertise, resulting in a deeper comprehension of complex medical issues and the development of comprehensive solutions.
Accelerated Innovation: The collaboration between researchers and clinicians allows for the seamless translation of scientific discoveries into clinical applications. Researchers can gain insights from the real-world experiences of clinicians and patients, ensuring that research efforts are aligned with practical medical needs.
- Patient-Centric Approach: Collaboration ensures that the patient remains at the center of research and healthcare efforts. By involving patients in discussions about treatment options, clinical trial designs, and research priorities, the resulting actions are more apt to meet the genuine requirements and desires of the individuals they aim to assist.
- Data Sharing and Integration: Collaborative efforts encourage the sharing of data across institutions and disciplines. This data sharing facilitates a more comprehensive analysis of medical information, which can uncover new patterns, trends, and potential treatment avenues.
B. Role of Patient Advocacy
- Empowerment and Education: Patient advocacy groups empower individuals by providing them with information about their medical conditions, treatment options, and rights as patients.8 This empowers individuals to make informed choices about their healthcare and actively participate in their treatment strategies.
- Research Prioritization: Patient advocacy groups often have a deep understanding of the challenges faced by patients. They can play a critical role in advocating for research funding and focusing research efforts on areas that have the greatest impact on patients' lives.3
- Policy Influence: These groups have the ability to influence healthcare policies, ensuring that patient perspectives are considered in decision-making processes related to regulations, drug approvals, and access to treatments.10
- Raising Awareness: Patient advocacy groups raise awareness about specific diseases and medical conditions, reducing stigma and increasing public understanding. This can lead to increased support, funding, and resources for research and treatment.
- Participation in Clinical Trials: Patient advocacy groups often facilitate the recruitment of participants for clinical trials, ensuring that studies have a diverse and representative pool of participants. 13
Conclusion
I. INTRODUCTION Millions of individual worldwide are impacted by sickle cell disease, which is still a serious global health concern. The genetic mutation causing SCD results in the aberrant production of hemoglobin, resulting to a cascade of events that end in vaso-occlusive crises and multi-organ damage.1 Historically, management has focused on palliative measures to alleviate symptoms and complications. However, recent breakthroughs in biotechnology and molecular medicine have paved the way for innovative pharmacotherapeutic interventions that target the root causes of sickle cell illness.3 The characteristic feature of SCD is the transformation of usually malleable red blood cells into rigid, sickle-shaped cells, triggering vaso-occlusive crises, agonizing pain, and progressive organ damage. The burden of SCD extends globally, with millions affected and diverse clinical manifestations ranging from mild to severe.10 The relentless nature of SCD has fueled a fervent pursuit of more effective therapeutic interventions. The advent of the evolving pharmacotherapeutic landscape has rekindled optimism within the medical community and among patients alike. Recognizing the inadequacies of traditional management approaches, researchers have embarked on a journey to target the underlying molecular intricacies of SCD.2 This review aims to navigate this journey, delving into the burgeoning realm of treatment strategies that hold the promise of transforming the outlook for SCD patients. The subsequent parts explain delve into the historical context of SCD treatment, dissect the mechanisms and potentials of novel therapies, and delve into the multifaceted challenges and opportunities that lie ahead.11 The Current analysis of the developing pharmacotherapeutic landscape, this review aim to offer an understanding of the monumental shifts underway in the pharmacotherapeutics of SCD.4 A. Traditional Approaches and Limitations The drug hydroxyurea is a medication that has been primarily used in the treatment of hematological disorders, most notably sickle cell disease and several forms of cancer.5 It exerts its therapeutic agents by as we approach the dawn of a new era in sickle cell disease management, our investigation not only clarifies state of the interventions but also emphasizes the critical importance of adapting and advancing treatment modalities.8 Its effect on DNA synthesis and cell division, which leading to the inhibition of cell proliferation. The clinical outcomes and mechanism of action of hydroxyurea in sickle cell disease are as follows: B. Mechanism of Action Hydroxyurea inhibiting the enzyme ribonucleotide reductase, which is involved in the conversion of ribonucleotides to deoxyribonucleotides. Because of this inhibition, fewer reduces the availability of deoxyribonucleotides, which are necessary for DNA synthesis and repair.15 Cell division is disrupted, leading to reduced production of red blood cells with abnormal shapes (sickle cells) in sickle cell disease. One type of haemoglobin that is specific to foetal tissue is called foetal haemoglobin (HbF) that normally present in newborns but diminishes as a person grows older. The molecular makeup of fetal hemoglobin differs from that adult hemoglobin and is less likely to forming the characteristic rigid (hard) & sickle-shaped cells.14 Fig: 1 Pathogenesis of sickle cell disease II. CLINICAL OUTCOMES Hydroxyurea has been linked with several beneficial clinical outcomes, such as: 1) Reduced Pain Crises: Hydroxyurea has been demonstrated to lessen the occurrence and intensity of painful vaso-occlusive incident, a characteristic feature of sickle cell disease.20 These crises occur due to the blockage of blood vessels by sickle-shaped red blood cells, leading to organ damage and discomfort. 2) Improved Anemia: Hydroxyurea can alleviate the inhance in hemoglobin levels and a reduce in reticulocyte count (immature red blood cells), addressing the anemia commonly seen in sickle cell disease.2 3) Decreased Incidence of Acute Chest Syndrome: Acute chest syndrome, a severe complication of sickle cell disease marked by chest discomfort, fever, and respiratory issues, has been linked to a lower likelihood in patients undergoing hydroxyurea treatment.7 4) Prevention of Stroke: Among pediatric individuals diagnosed with sickle cell disease, hydroxyurea has shown promise in reducing the risk of stroke. A. Challenges and Limitations of Hydroxyurea Therapy Despite its benefits, hydroxyurea therapy also comes with several challenges and limitations: 1) Variable Response: The response to hydroxyurea varies among patients with sickle cell disease. The level of increase in HbF and the clinical benefits can vary from person to person.16 2) Long-Term Safety: The long-term safety of hydroxyurea use is still a topic of ongoing research.11 Concerns have been raised about potential risks, include bone marrow suppression and heightened susceptibility to specific cancers. 3) Adherence: Hydroxyurea is a daily oral medication, and adherence to the treatment regimen can be challenging for some patients, especially considering the potential for side effects.13 4) Monitoring: Regular monitoring is required to adjust the dosage and assess the treatment\'s effectiveness and safety. This can be burdensome for patients and healthcare providers. 5) Pregnancy and Fertility: The use of hydroxyurea during pregnancy raises concerns about potential risks to both the mother and the developing fetus. It can also affect fertility in both men and women.21 6) Limited Availability: In some regions, access to hydroxyurea might be limited, preventing patients from benefiting from this treatment. Table: -1 A clear distinction between the difficulties and constraints of employing hydroxyurea treatment for sickle cell disease. S.No Challenges of Hydroxyurea Limitations of Hydroxyurea Therapy 1. Patient Adherence: Requires long-term commitment and consistent use. Daily medication can be challenging. Individual Response: Effectiveness varies among patients. Some patients may not respond well. 2. Side Effects: Typical adverse effects encompass nausea, fatigue, and myelosuppression, while a subset of patients may encounter more severe reactions. Cytotoxicity: Hydroxyurea is a cytotoxic drug, potentially affecting healthy cells along with sickle cells. 3. Dose Adjustment: Finding the right dosage requires careful monitoring and adjustment. Underdosing may be ineffective, while overdosing can lead to toxicity. Renal Impairment: Hydroxyurea is excreted by the kidneys, and caution is needed in patients with renal impairment. 4. Fertility Concerns: Potential impact on fertility, which can be a concern for some patients. Discussing family planning is crucial. Risk of Secondary Malignancies: Long-term usage could potentially elevate the risk of developing secondary malignancies. III. DISEASE-MODIFYING THERAPIES: These therapies aim to address various aspects of the disease and improve the well-being of individuals with sickle cell disease and elevate their quality of life. Let\'s briefly discuss each of these therapies: 1) Voxelotor: Voxelotor is a medication that works by increasing the levels of fetal hemoglobin (HbF) in individuals with sickle cell disease. HbF is a type of hemoglobin that is normally present in newborns and aids in preventing the formation of red blood cells from becoming misshapen and causing the characteristic sickling seen in SCD.8 By increasing HbF levels, voxelotor helps to reducing the formation of sickled red blood cells and the associated complications. 2) Crizanlizumab: Crizanlizumab is a monoclonal antibody that targets adhesion molecules involved in the vaso-occlusive process in sickle cell disease. Vaso-occlusion happens when sickled red blood cells aggregate and obstruct blood vessels, resulting in pain and organ damage.22 By inhibiting these adhesion molecules, crizanlizumab strives to lower the occurrence of vaso-occlusive crises and associated severe pain. 3) Luspatercept: Luspatercept is a medication primarily used to manage anemia in patients with certain types of blood disorders, including SCD.10 Anemia is a frequent side effect of SCD that can cause to fatigue and other health problems. Luspatercept works by promoting red blood cell production, which can help alleviate anemia and improve overall well-being in individuals with SCD.9 A. Emerging Frontiers: Gene Therapy and Editing In recent years, gene therapy and editing have become prominent as promising frontiers in the field of medical science, contribution to the potential treatment for a broad spectrum of genetic disorder. Two prominent approaches in this field are lentiviral-based gene therapy and CRISPR-Cas9 technology.25 These methods hold the potential to correct genetic mutations and alleviate the underlying causes of various diseases, particularly those caused by single-gene defects. B. Lentiviral-Based GENE Therapy Lentiviral-based gene therapy involves using modified lentiviruses to deliver functional copies of genes into the patient\'s cells. This allows the corrected gene to be permanently integrated into the patient\'s cells, theoretically providing a lasting therapeutic effect.22 This approach has shown success in correcting genetic mutations responsible for diseases like severe combined immunodeficiency (SCID) and certain forms of inherited blindness. By introducing functional copies of the mutated gene into the patient\'s cells, researchers aim to restore the normal cellular functions and alleviate disease symptoms. However, challenges remain, such as ensuring precise targeting of the correct cells, avoiding unintended insertions of the corrected gene, and managing potential immune responses triggered by the viral vectors. C. Crispr-Cas9 Technology CRISPR-Cas9 technology has been transformed the field of genetic editing. It is an accurate and flexible instrument that enable researchers to precisely target specific genes and modify particular gene within the DNA of an organism and make precise modifications. Regarding genetic illnesses, CRISPR-Cas9 can be used to edit the DNA sequence, either by correcting a mutation, introducing a functional gene, or disrupting a harmful gene.16 For instance, in the case of Beta-thalassemia, a genetic condition marked by impaired ?-globin production, CRISPR-Cas9 can be employed to precisely edit the ?-globin gene, restoring normal hemoglobin production and potentially curing the disease. D. Promising Results and Challenges Both lentiviral-based gene therapy and CRISPR-Case technology has demonstrated promising results in preclinical and clinical studies. Successful treatments have been reported for some genetic disorders, offering hope for patients and their families.17 However, challenges persist. Off-target effects, where genetic editing occurs at unintended locations, remain a concern with CRISPR-Cas9. Ensuring the safety and efficacy of these therapies in the long term is a critical aspect of their development. Additionally, ethical considerations surround the genetic modification of human embryos and germline editing, requiring careful ethical deliberation and oversight. IV. MANAGING PAIN AND COMPLICATIONS Pain Management Strategies for Vaso-Occlusive Crises: Vaso-occlusive emergencies are one of the trademark characteristic of sickle cell illness and can cause intense pain. These episodes happen when blood vessels are obstructed by sickle-shaped red blood cells, resulting in diminished blood circulation and harm to tissues.23 Effective pain control is essential for enhancing of life for people with SCD during these crises. Several approaches include: 1) Hydration: Staying well-hydrated helps prevent the clumping of sickle cells and can decrease the frequency and severity of vaso-occlusive episodes. 2) Pain Medications: Pain relievers are commonly used to manage pain during crises. Non-opioid analgesics such as nonsteroidal anti-inflammatory drugs (NSAIDs) like ibuprofen suitable for mild to moderate management. Opioid medications are typically reserved for severe pain and advisable to utilize them with the supervision of a medical expert. 19 3) Hydroxyurea: This medication promotes the synthesis of fetal hemoglobin, thus inhibiting the sickling of red blood cells. It can decrease the frequency and severity of vaso-occlusive episodes. 4) Blood Transfusions: In extreme circumstances, blood transfusions may be necessary to improve oxygen delivery to tissues and decrease the number of sickle cells. 5) Supplemental Oxygen: Administering oxygen can help improve tissue oxygenation and reduce pain during crises. A. Non-Opioid Analgesics and Alternative Approaches: Given the concerns about opioid addiction and side effects, non-opioid analgesics and alternative approaches are being explored to manage pain in SCD patients: 1) Non-NSAID Pain Relievers: Acetaminophen (paracetamol) is a substitute for NSAIDs in the treatment of agony in SCD patients. It\'s crucial to remember that NSAIDs can potentially worsen kidney function in some SCD patients.8 2) Nerve Blocks: Local anesthetic nerve blocks can offer specific pain relief for specific painful areas. 3) Physical Therapy: Techniques such as massage, stretching, and exercise can help manage reduce discomfort and increase mobility. 4) Cognitive Behavioral Therapy (CBT): CBT can assist individuals in managing their pain through relaxation techniques, mental imagery, and mindfulness.11 5) Mind-Body Practices: Practices like yoga and meditation can help individuals manage pain by promoting relaxation and reducing stress. B. Addressing Long-Term Complications of SCD: Sickle cell disease can result in a variety of long-term difficulty that necessitate continuous management: 1) Stroke Prevention: Individuals with SCD are at a greatest risk of stroke. Regular blood transfusions and the use of medications like hydroxyurea can help reduce this risk. 2) Organ Damage: SCD can cause harm to a number of various organs, including the kidneys, lungs, and liver. Regular medical check-ups are essential to monitor and manage these complications.17 3) Infection Prevention: SCD patients are more susceptible to infections. Vaccinations, prophylactic antibiotics, and prompt treatment of infections are important preventive measures. 4) Regular Monitoring: Routine blood tests and imaging are crucial for monitoring organ function and detecting potential complications early. 5) Pulmonary Hypertension Management: Pulmonary hypertension is a serious complication in some individuals with SCD. Medications and lifestyle changes may be prescribed to manage this condition.19 V. PATIENT-CENTERED CARE AND MULTIDISCIPLINARY APPROACHES Patient-centered care has gained significant importance in healthcare, emphasizing the need to focus on the individual\'s preferences, needs, and values when making decisions about their health. This approach is particularly crucial in the context of individuals with the complex genetic disease known as sickle cell disease that can have a wide range of physical, emotional, and social impacts. Holistic Care for SCD Patients: Shifting towards holistic care means examining the patient as a whole, rather than just focusing on their medical illness. For SCD patients, this involves recognizing that their well-being is influenced by various factors beyond their physical symptoms.24 This includes their emotional state, social support system, lifestyle, cultural background, and more. Holistic care for SCD patients involves not only treating the physical symptoms of the disease but also addressing their psychological and emotional needs, as well as their overall quality of life.1 Comprehensive Care Teams: Managing SCD effectively requires a multidisciplinary approach involving a team of healthcare professionals with diverse expertise. This team could include hematologists, nurses, pain specialists, social workers, psychologists, genetic counselors, nutritionists, and others.16 Mental Health Support: A patient’s mental health may suffer as a result of having a chronic illness such as SCD. Chronic pain, frequent hospital visits, and the uncertainty of the disease\'s progression can cause to anxiety, depression, and other psychological issues. Integrating mental health support into the care plan is essential. Mental health professionals can help patients and their families cope with the emotional and psychological impact of the disease, develop strategies to manage stress and anxiety, and improve overall mental well-being.20 Patient Education and Empowerment: Empowering SCD patients with knowledge about their condition is crucial. When patients understand their disease and treatment options, they can actively engage in making decision about their care. Education helps patients manage their symptoms, adhere to treatment plans, and recognize when they need medical attention.15 Additionally, involving patients in their care can foster a sense of control and enhance their encounter with the healthcare system as a whole. Cultural Sensitivity: SCD affects individuals from various cultural backgrounds, and cultural factors can affect how individuals view and treat their illness. Healthcare professionals should be aware of cultural difference, practices, and values can impact treatment decisions and communication. Incorporating cultural competence into care plans can improve patient trust, adherence, and outcomes.5 VI. CHALLENGES AND FUTURE DIRECTIONS 1) Accessibility and Affordability of Novel Therapies: Developing new and innovative therapies often involves significant research and development costs. As these therapies become available, ensuring they are accessible and affordable for a wide range of patients is a challenge. Regulatory agencies, governments, and healthcare organizations need to work together to balance the need for fair pricing with the financial sustainability of healthcare systems. 2) Long-term Safety and Monitoring Considerations: As new therapies are introduced, it\'s crucial to monitor their long-term safety and effectiveness. Some therapies might have unforeseen side effects or interactions that only become apparent after extended use. Robust post-market surveillance systems and real-world data collection are essential to identify and address any safety concerns that may arise. 3) Individualized Treatment Approaches Based on Genetic Factors: The field of personalized medicine is rapidly advancing, and genetic factors play a significant role in how individuals respond to treatments. Tailoring therapies to a patient\'s genetic makeup can enhance their efficacy and minimize adverse effects. However, implementing individualized treatment approaches on a large scale requires advancements in genetic testing technologies, data analysis, and regulatory frameworks to ensure privacy and ethical considerations. 4) Collaborative Research and Patient Advocacy: Driving Advancements in Healthcare In recent years, the landscape of medical research and healthcare has evolved significantly, placing a stronger emphasis on collaboration between researchers, clinicians, and patients. This collaborative approach has proven to be instrumental in driving advancements, improving patient outcomes, and accelerating the development of innovative treatments and therapies. A. Importance of Collaboration Cross-disciplinary Insights: Collaboration brings together experts from various fields, such as biomedical research, clinical medicine, engineering, and social sciences. This multidisciplinary approach fosters the exchange of diverse perspectives and expertise, resulting in a deeper comprehension of complex medical issues and the development of comprehensive solutions. Accelerated Innovation: The collaboration between researchers and clinicians allows for the seamless translation of scientific discoveries into clinical applications. Researchers can gain insights from the real-world experiences of clinicians and patients, ensuring that research efforts are aligned with practical medical needs. 1) Patient-Centric Approach: Collaboration ensures that the patient remains at the center of research and healthcare efforts. By involving patients in discussions about treatment options, clinical trial designs, and research priorities, the resulting actions are more apt to meet the genuine requirements and desires of the individuals they aim to assist. 2) Data Sharing and Integration: Collaborative efforts encourage the sharing of data across institutions and disciplines. This data sharing facilitates a more comprehensive analysis of medical information, which can uncover new patterns, trends, and potential treatment avenues. B. Role of Patient Advocacy 1) Empowerment and Education: Patient advocacy groups empower individuals by providing them with information about their medical conditions, treatment options, and rights as patients.8 This empowers individuals to make informed choices about their healthcare and actively participate in their treatment strategies. 2) Research Prioritization: Patient advocacy groups often have a deep understanding of the challenges faced by patients. They can play a critical role in advocating for research funding and focusing research efforts on areas that have the greatest impact on patients\' lives.3 3) Policy Influence: These groups have the ability to influence healthcare policies, ensuring that patient perspectives are considered in decision-making processes related to regulations, drug approvals, and access to treatments.10 4) Raising Awareness: Patient advocacy groups raise awareness about specific diseases and medical conditions, reducing stigma and increasing public understanding. This can lead to increased support, funding, and resources for research and treatment. 5) Participation in Clinical Trials: Patient advocacy groups often facilitate the recruitment of participants for clinical trials, ensuring that studies have a diverse and representative pool of participants. 13
References
[1] Torres L, Conran N. Emerging pharmacotherapeutic approaches for the management of sickle cell disease. Expert opinion on pharmacotherapy. 2019 Jan 22;20(2):173-86. [2] Hankins J, Aygun B. Pharmacotherapy in sickle cell disease–state of the art and future prospects. British journal of haematology. 2009 May;145(3):296-308. [3] Telen MJ. Developing new pharmacotherapeutic approaches to treating sickle?cell disease. ISBT science series. 2017 Feb;12(1):239-47. [4] Neville AK, Panepinto JA. Pharmacotherapy of sickle cell disease. 18th Expert Committee on the Selection and Use of Essential Medicines. 2011 Sep 28;1(1):1-5. [5] A Neville K, A Panepinto J. Pharmacotherapy of sickle cell disease in children. Current Pharmaceutical Design. 2015 Dec 1;21(39):5660-7. [6] Ataga KI, Orringer EP, Styles L, Vichinsky EP, Swerdlow P, Davis GA, DeSimone PA, Stocker JW. Dose?escalation study of ICA?17043 in patients with sickle cell disease. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy. 2006 Nov;26(11):1557-64. [7] Jaja C, Patel N, Scott SA, Gibson R, Kutlar A. CYP2C9 allelic variants and frequencies in a pediatric sickle cell disease cohort: implications for NSAIDs pharmacotherapy. Clinical and translational science. 2014 Oct;7(5):396-401. [8] Lunzer MM, Yekkirala A, Hebbel RP, Portoghese PS. Naloxone acts as a potent analgesic in transgenic mouse models of sickle cell anemia. Proceedings of the National Academy of Sciences. 2007 Apr 3;104(14):6061-5. [9] Shaiova L, Wallenstein D. Outpatient management of sickle cell pain with chronic opioid pharmacotherapy. Journal of the National Medical Association. 2004 Jul;96(7):984. [10] Bhagat VM, Baviskar SR, Mudey AB, Goyal RC. Poor health related quality of life among patients of sickle cell disease. Indian journal of palliative care. 2014 May;20(2):107. [11] Cieri?Hutcherson NE, Hutcherson TC, Conway?Habes EE, Burns BN, White NA. Systematic review of L?glutamine for prevention of vaso?occlusive pain crisis in patients with sickle cell disease. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy. 2019 Nov;39(11):1095-104. [12] Claudio AM, Foltanski L, Delay T, Britell A, Duckett A, Weeda ER, Bohm N. Antibiotic use and respiratory pathogens in adults with sickle cell disease and acute chest syndrome. Annals of Pharmacotherapy. 2019 Oct;53(10):991-6. [13] Kassim AA, DeBaun MR. The case for and against initiating either hydroxyurea therapy, blood transfusion therapy or hematopoietic stem cell transplant in asymptomatic children with sickle cell disease. Expert opinion on pharmacotherapy. 2014 Feb 1;15(3):325-36. [14] Ballas SK, Darbari DS. Review/overview of pain in sickle cell disease. Complementary Therapies in Medicine. 2020 Mar 1;49:102327. [15] Ataga KI. Novel therapies in sickle cell disease. ASH Education Program Book. 2009 Jan 1;2009(1):54-61. [16] Houwing ME, De Pagter PJ, Van Beers EJ, Biemond BJ, Rettenbacher E, Rijneveld AW, Schols EM, Philipsen JN, Tamminga RY, van Draat KF, Nur E. Sickle cell disease: clinical presentation and management of a global health challenge. Blood reviews. 2019 Sep 1;37:100580. [17] Bond K, Campbell C, Lanzkron S, Haywood C, Carroll P, McCauley L, Buenaver L, Haythornthwaite J. Situational catastrophizing mediates laboratory pain responses in sickle cell disease patients. The Journal of Pain. 2013 Apr 1;14(4):S34. [18] Amanzholova DT. PROBLEMS AND PROSPECTS OF PHARMACOTHERAPY OF SICKLE CELL DISEASE. ??? 51 ? 33. 2014:18. [19] Bennett N, Mulhall J. Sickle cell disease status and outcomes of African-American men presenting with priapism. The journal of sexual medicine. 2008 May;5(5):1244-50. [20] McCauley SR, Pedroza C. Event-based prospective memory in children with sickle cell disease: Effect of cue distinctiveness. Child Neuropsychology. 2010 Apr 21;16(3):293-312. [21] Campbell CM, Moscou-Jackson G, Carroll CP, Kiley K, Haywood Jr C, Lanzkron S, Hand M, Edwards RR, Haythornthwaite JA. An evaluation of central sensitization in patients with sickle cell disease. The Journal of Pain. 2016 May 1;17(5):617-27. [22] Doss JF, Jonassaint JC, Garrett ME, Ashley-Koch AE, Telen MJ, Chi JT. Phase 1 study of a sulforaphane-containing broccoli sprout homogenate for sickle cell disease. PloS one. 2016 Apr 12;11(4):e0152895. [23] Hutcheson J. Bone infarction in sickle cell disease. American Journal of Roentgenology. 1979 Jun 1;132(6):1025-6. [24] Works T, Jones S, Grady J, Andemariam B. Traumatic exposure history as a risk factor for chronic pain in adult patients with sickle cell disease. Health & Social Work. 2016 Feb 1;41(1):42-50. [25] Jenerette CM, Murdaugh C. Testing the theory of self?care management for sickle cell disease. Research in nursing & health. 2008 Aug;31(4):355-69.
Copyright
Copyright © 2024 Nancy ., Harmanjot Kaur, Dr. Shivam Choudghal, Pardeep Kumar. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET64436
Publish Date : 2024-10-02
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online