

Ijraset Journal For Research in Applied Science and Engineering Technology
Identification of the Novel Genes Associated with the Inflammatory Bowel Disease (IBD) using Genome Wide Expression Sequencing Data Set
Authors: Payal Priyadarshini, Ankur Chaurasia
DOI Link: https://doi.org/10.22214/ijraset.2024.62654
Certificate: View Certificate
Abstract
Inflammatory Bowel Disease (IBD) is a prevalent chronic condition worldwide, representing a significant global health challenge due to its varying prevalence rates across different regions. Comprising Crohn’s disease (CD) and ulcerative colitis (UC), IBD entails chronic inflammation of the gastrointestinal tract. Crohn’s disease can manifest in any part of the gastrointestinal tract. Genomics serves a pivotal role in elucidating the fundamental genetic underpinnings of IBD, facilitating the diagnosis of patients with exceedingly rare disease variants. Next-generation sequencing technologies have been extensively employed to analyse IBD datasets on a global scale, while targeted sequencing methods have enabled researchers to identify patients and genes associated with rare disorders within the spectrum of IBD. This study utilizes microarray sequencing to derive insights from selected datasets. Data sourced from the NCBI-GEO public database encompassing microarray/gene profiles provided the gene expression profiles for dataset GSE6731, containing samples representing Normal, UC, and CD conditions. Following successful extraction of expression values, 9156 differentially expressed genes were identified for each condition—Crohn’s disease, ulcerative colitis, and combined IBD—as specified in the design matrix. Furthermore, a correlation analysis was conducted to elucidate the relationship between these genes and their respective control origins, along with inflammatory biomarkers in the cells and tissues. Through the integration of microarray sequencing and correlation analyses, this research aims to deepen our understanding of the molecular mechanisms underlying IBD pathogenesis. By identifying differentially expressed genes and their associations with specific disease phenotypes, this study contributes valuable insights that may inform the development of targeted therapies and precision medicine approaches for managing IBD and its variants. This study underscores the significance of genomics in unravelling the complexities of IBD, offering insights into potential therapeutic targets and paving the way for personalized treatment strategies in the management of this multifaceted disease.
Introduction
I. INTRODUCTION
The inflammatory bowel disease (IBD) is one of the most popular yet chronic diseases in the world and is considered a global health concern with its significantly varying rates in different parts of the world. This term is used for two conditions, i.e., Crohn’s disease (CD) and ulcerative colitis(UC), which is a chronic gastrointestinal tract inflammation can be visualised in figure 1. Crohn’s disease can occur in any part of the gastro-intestinal tract (Jairath & Feagan, 2020). Adding to the catastrophic effect on the lower section of the small intestine, CD can develop in parts of the digestive tract such as the large intestine, stomach, oesophagus, or even mouth. The ulcerative colitis affects the large bowel region and is characterised with blood in the faeces, intense discomfort, and diarrhoea (Seyedian et al., 2019).
Figure 1: A visual representation of different conditions of Inflammatory Bowel Disease showcasing control condition (extreme left), Crohn’s disease(middle) and Ulcerative Colitis(extreme right) both of which have been classified as chronic IBD and which cause digestive disorders and inflammation in the gastrointestinal tract (Seyedian et al., 2019)and have overlapping clinical features and complex pathophysiologies.
The inflammatory bowel disease was more prevalent in western parts of the world during the 19th and early 20th centuries and was found in isolated parts of the United Kingdom and northern Europe (Actis et al., 2019). Globally there are around 10 million cases (Kirsner, 2001) from which 1.4:1.3 ratio of the patients were males and females respectively. The incidents of IBD have now spread across the south Asian countries like China, Malaysia, India, Singapore etc, with an average of overall estimation of 1.4 million cases according to the studies standing on the 2nd position after the United States with 1.64 million cases (Kedia & Ahuja, 2017).
According to the survey, the mortality rate of IBD patients is observed to be 20-26.55% in South- Asian countries, and for the United Kingdom, the rate was 17.1% per 1000 IBD patients (Burisch et al., 2013). The 70% CD sufferers and 30% UC patients will require surgery at least once in their lifetime, the occurrence of UC is twice as that of CD in Asian countries, and is distinctively considered a rare disease, whereas, in Australia it was observed that this ratio is reversed (Latella, 2012). Characteristics like, Genetics, geomorphological changes, environment, diet, microbiome, climate, age, smoking history contribute towards the chronic inflammation of IBD. It was observed that the most cases are from the age groups of 20-39 and 60 to 79 years in the western countries and 36–38-year age groups in India (Mulder et al., 2014).
IBD affects people during their peak years of productivity and has been linked to serious complications and functional capacity loss, as well as the burden of expensive and time-consuming therapy. IBD in India is equally invasive as in the West, demanding a similar therapeutic strategy (Mulder et al., 2014). The number of patients with IBD is rapidly increasing. Researchers are looking for novel medicines to remove the disease and reduce its effects. IBD is a severe condition with serious consequences for the patients. It impacts patients' overall well-being and standard of life (Cheng et al., 2020).
Significant progress has been made in several omics levels, including the genome, transcriptome, proteome, metabolome, and microbiome (Loddo & Romano, 2015). Nonetheless, despite this progress, several studies continue to evaluate those omics independently, without considering their complex relationships in health or sickness. As a result of a single focal point, one omics strategy applied at a time can only explain one feature of any complicated disease (Kumar et al., 2019). Further, genomics and proteomics are among the most widely used data as they are easily available on online platforms and the technologies to process the data have been evolving and cost effectively utilised worldwide (Assadsangabi et al., 2019).
Genomics plays a crucial role in the identification of the basic genetic markup of the inflammatory bowel disease(IBD) further diagnosing patients with extremely rare variants of the disease. Apart from its diagnostic abilities, genomics provide insight over Mendelian disease-associated IBDs and can also improve the clinical classification of people suffering from IBD(Ahmad et al., 2006). It gives a predictive analysis to the response of any therapy, contributing to pathway specific therapies and bone-marrow transplantation hereby, giving an understanding about the adverse side-effects due to clinical history of their past diseases. According to previous studies for genomics study, GWAS(genome-wide association studies) has widely examined the aid of genetic variation and next generation sequencing technologies in the analysis of the datasets and detecting anomalies (Yamada et al., 2020). The potential of personalised medicine is also embarked through the interconnection of Genomic information and inflammatory bowel disease(de Lange et al., 2017). Exome sequencing or predominantly known as Genome sequencing is thought to be the most cost-effective way to examine big patient samples; low-coverage genome sequencing may serve as an interim step for genome sequencing for massive IBD samples (Loddo & Romano, 2015). Genome-wide scans have revealed persistent evidence of linkage to IBD3 (6p21.1-23), a region that includes the HLA complex, for both Crohn's disease and ulcerative colitis, with a number of replicated connections with disease susceptibility and phenotype emerging recently (Mahdi, 2015). The HLA complex, which has also been found to be identified in the analysis part of this paper, refers to the major histocompatibility complex, can be detected on chromosome 6p21.3. HLA genes generate particular cell surface proteins of antigen-representing cells that trigger a T-cell-mediated immune response. Based on prior research, HLA genes are strongly responsible for the development of IBD and are divided into two major groups: class I consisting of HLA A,B, and C, and class II genes with HLA DR, DQ, and DP (Kumar et al., 2019).
In IBD, developments in the field of molecular genetics and the development of genome-wide association studies enabled the identification of numerous candidate genes whose pathogenic mutations or single nucleotide polymorphisms are associated with an elevated risk of CD or UC (Fabian et al., 2023). Additionally, understanding the proteome's composition and changes provides insights into IBD pathogenesis as well as possible biomarkers of disease activity, mucosal repair, and tumour growth (Assadsangabi et al., 2019). Proteomics may ultimately relate genomic and transcriptome abnormalities to the phenotypic expression of IBD, allowing for a better understanding of the mechanisms underlying illness beginning and progression (Chan, 2016). Previously, the search for prospective biomarkers was restricted to a select list of proteins thought to be involved in IBD aetiology. Based on the study of a wide range of proteins in tissues, it appears to be a viable strategy for discovering new biomarkers (Titz et al., 2018). Modern proteomic techniques, such as mass spectrometry (MS) and the adoption of complicated bioinformatic studies, have made it possible to isolate and analyse a huge number of proteins, increasing the likelihood of discovering a possible new biomarker.
Improvements in omics sequencing technologies, which include: Whole-exome sequencing (WES), Targeted sequencing, Transcriptomics (RNA-Seq) and mass spectrometry, have opened up the implementation of molecular markers for diagnosis of omics datasets (Yamada et al., 2020). Additionally, along with the huge population of patients with polygenic IBD, numerous rare Mendelian illnesses can cause IBD-like intestinal inflammation. The words monogenic IBD and IBD like inflammation are increasingly utilised interchangeably in the literature, emphasising the commonality in histological characteristics but emphasising differing genetic architectures compared to polygenic IBD (Han et al., 2018).
There are several sequencing approaches, each of which provides unique insights into the disease's molecular mechanisms. The next generation sequencing technologies have been widely used to analyse IBD datasets across the globe, further the wide spectrum of targeted sequencing methods helped researchers to identify patients and genes with rare disorders related to Inflammatory Bowel Disease (Loddo & Romano, 2015). Several studies related to Genome sequencing methods have proven ground breaking insights on this particular disease. The data types of IBD play an informative and crucial role in the process analysis and are being widely obtained in the form of clinical data, genomic and transcriptomic data, imaging data, microbiome data, and universal survey data. The most commonly used data types are clinical and universal surveyed data, to obtain real-time conclusions and results (Fiocchi et al., 2023). Although the various molecular characteristics, biological markers, and therapeutic targets of IBD previously discovered have contributed significantly to its diagnosis and treatment, the biological complexity, outcome severity, and high metastasis of this complex disease necessitate further predictive and prognostic biomarker identification (Loddo & Romano, 2015).
In this paper, microarray analysis is utilised to obtain the desired results from the chosen datasets. Differential gene expression analysis of sick and healthy samples can reveal molecular pathways that are abnormal in IBD. A workflow for the same has been illustrated in figure 2. We significantly aim to process the equipped data, further design, and develop efficient and novel pipelines to study the genetic association with IBD and further investigate the genes linked to inflammatory markers.\
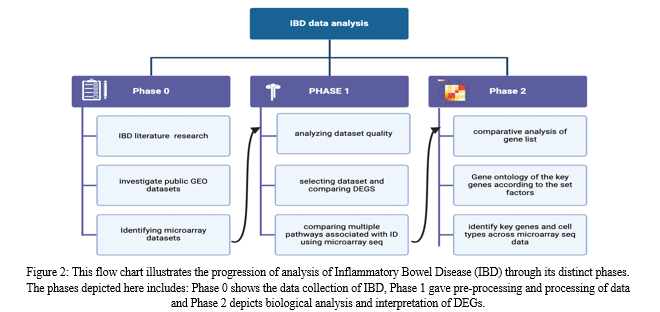
II. REVIEW OF LITERATURE
Inflammatory bowel disease (IBD) is a diverse set of illnesses defined by chronic inflammation of the gastrointestinal tract. Over time, advances in genomic technologies, notably genome-wide expression analysis, have yielded important insights into the molecular pathways driving IBD pathogenesis. This review will summarise the current literature on the use of genome-wide expression data to identify novel genes linked with IBD. A thorough search of databases such as PubMed, Scopus, and Web of Science was carried out to find relevant papers published in peer-reviewed journals. To locate pertinent studies, search terms including "inflammatory bowel disease," "genome-wide expression," "gene expressions profiling," and "novel genes" were utilised. This review comprised studies that used genome-wide expression data to uncover novel genes involved in IBD pathogenesis.
The literature review on the identification of novel genes using genome-wide expression data in inflammatory bowel disease (IBD) emphasises the diverse character of this approach to understanding the molecular pathways underlying IBD pathogenesis. Numerous studies have used genome-wide expression profiling to investigate gene expression patterns related with IBD, revealing a multitude of putative candidate genes involved in disease susceptibility, progression, and response to therapy. These studies have provided light on the intricate interaction of genetic predisposition, environmental variables, and immunological dysregulation in IBD. Notably, developments in high-throughput sequencing technologies have permitted the discovery of novel genes and pathways implicated in IBD pathogenesis, providing important insights into disease heterogeneity and personalised therapy methods. However, issues like as sample heterogeneity, data interpretation, and finding replication across varied populations continue to be important considerations in the search for strong and clinically relevant gene signatures for IBD. Future research projects that use integrated omics techniques and large-scale collaborative initiatives show promise for better understanding the genetic architecture of IBD and converting these insights into better diagnostic and therapeutic therapies.
While there are similarities between Crohn's disease and ulcerative colitis, there are also differences in the type and site of inflammatory alterations. The main difference between these two conditions is that ulcerative colitis is characterised by inflammation restricted to the large intestine, whereas Crohn's disease can cause inflammation anywhere in the gastrointestinal system. The frequency of Crohn’s disease is comparatively high in nations that are highly industrialised the incidence of this disorder is five in 100,000 people and its prevalence is estimated to be 30–50 of 100,000 persons in western countries. Crohn's disease affects the entire intestinal wall at the microscopic level, whereas ulcerative colitis is restricted to the gut's epithelial lining. Since the two diseases share comparable signs and symptoms, the diagnosis of one form of IBD over the other is frequently very difficult. As was previously mentioned, immunosuppressive medications and anti-inflammatory compounds play a major role in the medical treatment of inflammatory bowel disease (IBD). Citation9 Despite the availability of immunosuppressive therapies like methotrexate, azathioprine, and mercaptopurine, achieving a remission of IBD in patients remains a clinical challenge. Citation10 This class of therapeutics is very effective in reducing the extent of inflammation, but it comes with a wide range of side effects.
Epidemiological studies have highlighted rising incidence and prevalence rates worldwide, with notable geographic and ethnic variations. The aetiology of IBD involves a complex interplay between genetic predisposition, environmental stressors, dysregulated immune responses, abnormalities in the gut microbiota, and mucosal barrier dysfunction. Numerous susceptibility loci and gene variants linked to inflammatory bowel disease (IBD) have been found thanks to advancements in genomic research, which have also revealed possible treatment targets. Clinically, IBD manifests with a wide spectrum of symptoms, including abdominal pain, diarrhoea, rectal bleeding, weight loss, and extraintestinal manifestations, leading to substantial morbidity and impaired quality of life.
Diagnostic modalities encompass clinical evaluation, endoscopic assessment, radiological imaging, and laboratory testing, with histopathological confirmation being essential for definitive diagnosis. The goals of IBD treatment plans are to reduce symptoms, avoid complications, bring the illness into, and keep it out of remission, and enhance long-term results. Amino salicylates, corticosteroids, immunomodulators, and biologic agents that target particular cytokines or cell surface receptors are examples of conventional therapies. However, difficulties like as medication resistance, side effects, and disease relapse underline the need for customised and multidisciplinary care approaches. New therapeutic pathways that represent a paradigm shift towards precision medicine and comprehensive care include faecal microbiota transplantation, new biologics, small compounds, stem cell therapy, nutritional therapies, and psychosocial interventions.
Additionally, research activities focus on identifying the function of the gut microbiome, host-microbe interactions, epigenetics, metabolomics, and immune regulation in determining IBD pathogenesis and therapeutic responses. Collaborative initiatives, patient registries, and clinical trials play essential roles in expanding knowledge, promoting innovation, and improving patient outcomes in the field of IBD. Despite tremendous progress, unsolved issues continue regarding disease heterogeneity, predictive biomarkers, appropriate therapy algorithms, and techniques for attaining durable remission and mucosal healing. Future directions in IBD research comprise using big data analytics, artificial intelligence, patient-centered outcomes research, and precision medicine approaches to solve these issues and usher in a new era of personalized and tailored care for patients with IBD.
Research on inflammatory bowel disease (IBD) has recently placed a great deal of emphasis on finding new genes through expression profiling methods. Researchers are able to thoroughly examine gene expression patterns in IBD patients in comparison to healthy controls or other disease states thanks to genome-wide expression profiling, which is made possible by next-generation sequencing and microarray technology. These investigations have uncovered a number of potential genes linked to the severity, propensity for the disease, and response to therapy, as well as insightful information about the dysregulated molecular pathways underpinning IBD pathogenesis. Gene expression investigations in IBD have yielded several important discoveries.
Firstly, these studies have brought to light the significance of immunological dysregulation and pathways connected to inflammation in the pathophysiology of disease. Numerous studies have consistently linked genes related to leukocyte trafficking, cytokine signalling, antigen presentation, innate and adaptive immunological responses, and IBD pathogenesis.
Significant variation in gene expression patterns across IBD patients has also been found using expression profiling, which is consistent with the variety of clinical characteristics and disease courses seen in this patient population. Subgroup analysis based on treatment response (e.g., biologic responders vs. non-responders), disease behaviour (e.g., structuring vs. penetrating CD), disease location (e.g., ileal vs. colonic involvement), and disease phenotype (e.g., CD vs. UC) has contributed to our understanding of the molecular basis of disease subtypes.
Furthermore, mechanistic insights into the impact of treatment interventions on the molecular hallmark of IBD have been obtained through gene expression investigations. Treatment with biologic drugs, for instance, that target certain cytokines or cell surface receptors has been demonstrated to alter patterns of gene expression linked to inflammation, mucosal healing, and tissue repair, thereby offering clues to the therapeutic mechanisms of these drugs. Even with these developments, finding and validating reliable gene signatures for IBD still presents difficulties. Reproducibility and generalizability of results across research are severely hampered by problems such sample heterogeneity, inconsistent experimental procedures, lack of standardised data analysis pipelines, and small sample sizes.
In the future, integrative methods that combine gene expression profiling with other omics technologies—such as proteomics, metabolomics, and genomics—may be able to clarify the intricate molecular pathways that underlie the pathophysiology of IBD. To validate candidate genes, aggregate large-scale datasets, and convert research findings into clinically useful insights for the diagnosis, prognosis, and treatment of IBD, collaborative efforts, multi-center consortia, and data sharing programmes are critical.
III. METHODOLOGY
A. Analysis Optimization
Inflammatory bowel disease (IBD) is a spontaneous, chronic condition with an unknown cause and an underlying hereditary risk. Recent genome-wide association research investigations have found over 200 IBD susceptibility loci, however the underlying causes of IBD remain unknown (Frenkel et al., 2019). Genome-wide expression analysis promises to contribute to the functional annotation of genomes and has already provided a wealth of differentially expressed data.
Expression data from each experiment must first be normalised to account for systematic experimental variation, including unequal dye incorporation and detection efficiencies. For comparison between experiments, data is often first filtered to select a subset or to exclude genes for which there is much missing data (Palmieri et al., 2015). A distance matrix must then be chosen, which determines how we measure similarity between gene-expression patterns. The genes and experiments are subsequently grouped through different computational approaches. Each step can influence how the expression data are grouped (Chan, 2016). Analysis of Genome-wide expression data includes: Data collection depending on the availability, Feature extraction, Quality control metrics, Normalisation, Differential expression analysis, additionally Biological interpretation of the results and principal component analysis depending on the desired result has been performed for this project.
B. Acquisition of Data of Gene Expression Profiles
The data for the analysis of IBD samples usually starts from a wet laboratory technique where the biological samples like tissue biopsies and blood samples are the point of initiation, which are obtained from both healthy(control) and IBD patients. Following this process, isolation of DNA or RNA extracts further converting them and purifying them. Specific labelling is necessary for the preparation of microarray slides. Hence, fluorescent labels are attached to the DNA. Since two or more samples are compared in a genome sequencing experiment, each sample is labelled with a different coloured fluorescent dye. Thereby, labelled DNA or cDNA samples are applied to a slide containing hundreds to millions of distinct DNA probes, each related to a gene or genomic area of interest. These samples then hybridise (bond) with the complementary sequences on the slide. Following hybridization, the slide is washed to eliminate any free or non-specifically bound DNA (Linggi et al., 2021). The slide is then scanned with a laser, which excites the fluorescent dyes attached to the samples. A scanner detects the emitted light and measures fluorescence intensity at each location. This intensity represents the amount of DNA or RNA in the original sample for each gene or region. This method allows researchers to examine gene expression levels, detect changes in genetic activity, and get insight into biological processes. The scanned image produces raw data in the form of intensities for each spot. Such raw files are extracted from databases available online in order to save time and efficiency hence provide a platform for summarised information about the datasets (Wang et al., 2022).
C. Pre-processing of data
Pre-processing steps are applied to remove background noise and normalise signal intensities, followed by quality control checks to ensure data reproducibility. Statistical analysis identifies differentially expressed genes (DEGs) between experimental groups, and functional enrichment analysis interprets DEGs in the context of biological pathways and processes (Federico et al., 2021). Validation of microarray findings using independent experimental approaches and follow-up studies further elucidate the functional relevance of identified DEGs in IBD pathogenesis and therapeutic targeting (Singh & Singh, 2020).
D. Normalisation of expressed values
Data normalisation is an important step in gene expression analysis. It entails modifying raw data to account for variables that prohibit direct comparisons of expression measures. Normalisation techniques involve suppositions regarding the experiment and the expression of genes. In microarray analysis, it seeks to eliminate variables that influence the measured gene expression levels (Olsen et al., 2016). This makes it easier to discern biological variations and compare levels of expression across slides. The whole purpose of the normalisation technique is to reduce systematic changes in the measured gene expression levels of two co-hybridized mRNA samples, making biological differences easier to discern, and to allow expression levels to be compared across slides. (Yang et al., 2002).
E. Principal Component Analysis
Principal components analysis (PCA) is a statistical technique for identifying the important variables in a multidimensional data set that explain observed variations.
It can be used to simplify multidimensional data processing and visualisation. It is a dimensionality reduction method that is commonly used to reduce the dimensionality of big data sets. It works by reducing a large collection of variables into a smaller one that retains most of the information from the larger set. PCA is formed for the basis of multivariate data analysis based on projection methods (Bjerrum et al., 2014).
The primary application of PCA in this context is the representation of a table of multivariate data as a smaller collection of variables for the purpose of identifying trends, leaps, clusters, and outliers. A principal component analysis (PCA) plot depicts the similarities between groups of samples in a dataset. Each point on a PCA plot reflects the relationship between an initial variable and the first and second main components (Ahmed et al., 2016). One of the two variables in a PCA analysis of DNA microarray outcomes could be the experiments or the genes. When genes are used as variables, the analysis generates a set of "principal gene components" that identify the characteristics of genes which most effectively explain the experimental responses they cause.
F. Differential Gene Expression analysis
Raw Affymetrix data (CEL files) were downloaded and translated into expression values, which were then corrected for background. Gene expression data were then normalised using the robust multiarray average technique. R Bioconductor is s systematic collection of code, documentation, and/or data used to do specific types of analysis, such as affy, cluster, and graph packages.It has been used to perform the extraction of cel files from the system and further perform the basic normalisation and extract the expression values of the 32 samples considered from the dataset.
G. Biological Interpretation
The biological interpretation of gene expression data can assist in determining whether differentially expressed genes (DEGs) are related with a specific biological process or molecular function and binding. Many of the approaches for visualising and understanding gene expression data are applicable to both microarray and RNA-seq research analysis. Gene enrichment analysis is a popular method for evaluating gene expression data that is based on functional annotation of DEGs.
Enrichment of biological pathways provided by KEGG, Ingenuity, Reactome, and WikiPathways. Gene set analysis (GSA), another name for functional enrichment analysis, is a popular technique for analysing high-throughput experimental data. GSA seeks to identify biological annotations that are over-represented in a set of genes when compared to a reference background. The Gene Ontology, which includes uniform annotations of gene products, is widely used for the purpose of biological interpretation. It works by comparing the frequency of individual annotations in the gene list (such as differentially expressed genes) to a reference list (often all genes on the microarray or in the genome).
IV. RESULTS
A. Sample Collection
The datasets for this paper were retrieved from the NCBI-GEO (Gene Expression Omnibus) a free public database of microarray or gene profile, and we obtained the gene expression profile of GSE6731-platform [HG_U95Av2] Affymetrix Human Genome U95 Version 2 Array for healthy, UC and CD samples of the dataset. High throughput sequencing data profiling of gene expression is included in the data. The present data for GSE6731 includes out of these 36 cases, 4 are controlled cases, while 32 are samples with adult inflammatory bowel disease (IBD). Majority of cases have Crohn’s disease i.e., 19 while 9 have ulcerative colitis. Although the age group has not been considered for the analysis for this paper, the number of cases for each dataset is summarised below in Table 1. The research data was collected and conducted at the Johns Hopkins University School of Medicine,specifically in the Department of Medicine, Baltimore, USA. The study was submitted on January 12, 2007, and last updated on December 13, 2018.
|
Data set Accession ID |
Type of the data (file format) |
Technology type (if any) |
Number of cases |
Reference |
|
GSE6731 |
Microarray( CEL files) |
in situ oligonucleotide |
36 samples (34 considered) |
Table 1: The following table consists of dataset used for a genome wide sequencing analysis, each containing control/non-inflammatory and inflammatory samples, where GSE6731 is taken from Affymetrix Human Genome U95 Version 2 Array, and dataset GSE9452 and GSE16879 taken from U133 Plus 2.0 Array
- GSE6731
The following dataset has been procured from NCBI GEO website where it matched the desired criteria of IBD conditions dataset. In order to perform Genome-wide sequencing of genes, cel files from the Affymetrix array are required. The expression data shows the variations in Crohn's and ulcerative colitis's genome-wide gene expression from endoscopic pinch biopsies. The research study compared active and inactive areas of UC and CD with infectious colitis and healthy controls. A total of 36 samples underwent unsupervised classification, revealing distinctive gene expression patterns among affected and unaffected IBD tissues, non-IBD colitis, and normal controls.
|
RAW FILE FORMAT |
SAMPLES NUMBERS |
CONDITIONS |
SHORTLISTED CONDITIONS |
|
celfiles |
36 |
Ulcerative colitis-9 |
Ulcerative colitis-9 |
|
|
|
Crohn’s disease-19 |
Crohn’s disease-19 |
|
|
|
Normal-4 |
Normal-4 |
|
|
|
Indeterminate colitis -2 |
|
|
|
|
INF(bacterial infectious colitis)-2 |
|
Table 2: the GEO dataset GSE6731 has .CEL format taken from Affymetrix Human Genome U95 Version 2 Array where the shortlisted conditions are normal, UC and CD which are in total 34 out of the 36 samples present in the original dataset.
B. Pre-processing of the Extracted Sample Files
Data preparation, processing and normalisation are among the first steps into the analysis of the first dataset GSE6731. In order to retrieve the expression values of the samples in IBD, the pre-processing of data was performed for the collection and the retrieval of DEGs afterwards. Then to retrieve the RAW file either we can use a set of R CODE or directly download and unzip the compressed version of the raw data file.
Once the RAW files have been decompressed the .cel files also have to be retrieved, i.e. unzipped in order to be read by the R studio console. Library affy has been used to perform the extraction of cel files from the system and further perform the basic normalisation and extract the expression values of the 32 samples of UC, CD and normal control are considered from the dataset.
C. Normalisation of Expressed Values
Readaffy function reads the cel files into the R environment and prepares it for normalisation, exprs and rma are both functions used for normalising the data. Successfully extracting the expression values of the samples, now QC must be performed on the normalised data to anomalies and outliers. To detect the accurate normalisation of the dataset, Figure 3 shows a boxplot plotted to visualise the normalisation of the expression values from the samples.
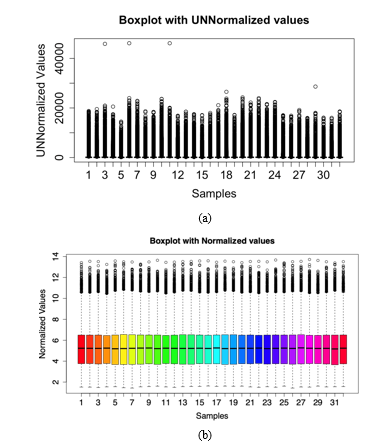
Figure 3: a Boxplot illustration to showcase data variance and normalisation in the samples of the dataset GSE6731. Samples are listed on the x-axis where 32 patient data is listed and the expression values are represented on the y-axis (a) A boxplot showing expression values of the data with unnormalized configuration, (b) Boxplot depicting normalised expression values after quality control of the data. The colour palette can be changed according to the user’s choice(note: here the palate has been set as “rainbow” to give a visually appealing aesthetic touch to the box plot).
Normalised values often show the variance of the samples showcasing the regulated genes through their expression values, this can be again visualised using a heatmap plot Figure 4 where the upregulated and downregulated genetic variance can be noticed using different colour themes often present in the Bioconductor package. Library (ComplexHeatmap) for this purpose has been utilised to illustrate the expression values of the normalised data and further the colorRampPalette command to make a combination of different colour schemes that are used to construct the heatmap.
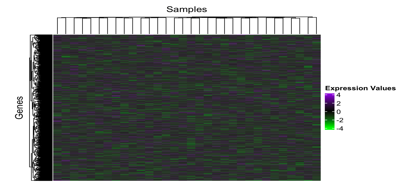
Figure 4: A heatmap constructed with the help of ComplexHeatmap package in R version 4.3.2, in the above graph, columns consist of the 32 samples and the associated regulated genes are somewhere represented in the rows. Due to the large number of expressed values here the map shows significant boxes overlapping with mild to high variance in the values. The Violet colour represents the Upregulated genes whereas the downregulated genes are shown in the green colour. The value ranges from +6 to -6 in the plot.
D. Principal Component Analysis
In the following dataset, a PCA is done using the expression values of Inflammatory Bowel Disease dataset to demonstrate any possible sample clustering within and variation between cohorts. It shows the correlation or variability of the samples from each other and gives a visual characterization to differentiate the normal vs diseased samples. In order to generate the plot, ggfortify library has been used in the R pipeline, further the pc data has to be generated where each sample contains the individual pc score using the expression values. The function autopilot has been used to generate a pc plot for and has been shown in the figure 5. The Principal Component 1 (PC1) is the first principal component extracted from the data which captures the direction of maximum variance in the data whereas Principal Component 2 (PC2) is the second principal component, orthogonal (perpendicular) to PC1, capturing the next highest amount of variance that is uncorrelated with PC1. The percentage numbers for each primary component reflect how much of the total variance is explained by that component. For example, if PC1 explains 55.59% of the variance, this means that when the data points are projected onto PC1, this single component accounts for 55.59% of the total variability in the data. In a similar manner if PC2 represents 11% of the variation, it means that PC2 accounts for an additional 11% of overall variability in the data set that PC1 does not explain. It is interpreted that "55.59%" for PC1 and "11.18%" for PC2 indicate that PC1 is the dominating component, accounting for the majority of the variation in the data. PC2 with a percentage that is lower (11.18%), detects less variation, but it provides additional information not described by PC1. When reading a PCA graphic, the axes (PC1 and PC2) do not correspond to the original variables in the dataset.
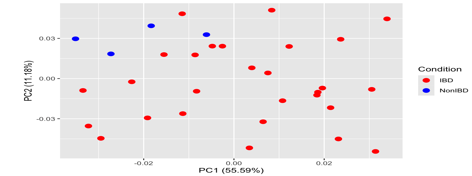
Figure 5: Principal component analysis performed on the IBD dataset GSE6731. The points here represent the samples and are coloured according to the subject cohort. Results are plotted according to the PC1 and PC2 scores from the pc data matrix generated using ggfortify library, with the percent variation explained by the respective axis where the PCA plot is demonstrating variation between IBD and non-IBD samples.The PC1 score on x axis demonstrates a 55.59% showing the maximum variance and the orthogonal PC2 score is 11.18% capturing the next highest amount of variance.
E. Differential Genes Expression analysis
Differential Gene Expression (DGE) searches for the genes with expression that varies in response to therapy or between groups. After successfully normalising our data the next step is to perform differentiation of the expressed genes or retrieve differentially expressed genes which will be further used for biological interpretation. We initiate by accessing the limma library, limma is an R/Bioconductor software package that provides an integrated solution for analysing data from gene expression experiments. It is a very popular package for analysing microarray and RNA-seq data. We construct a design matrix for the healthy control samples, Crohn's disease and Ulcerative colitis samples, and further make a contrast matrix of IBD vs Non IBD samples which consist of CD and UC. Depending on the type of genes you wish to retrieve you may construct different contrast matrices. For the analysis, CD - normal, UC - normal and Combined - normal conditions were taken into account.
After the formation of the matrix, we further proceed with ranking genes in order of evidence for differential expression using the function eBayes. This step is critical for stabilising variance estimates, especially when there are a limited number of samples to analyse or genes with low expression. "Fit" refers to outcomes of fitting a linear model to gene expression data. This linear model have been fitted with a function like lmFit() from the limma package for microarrays. To compute the contrasts indicated in the contrastMatrix, we used the fit() function from the limma package (or a comparable function from another package). It takes the fitted model (fit) and the contrast matrix as inputs and computes the desired contrasts. After this procedure, we need to produce a top table of the conditions used in the contrast matrix we constructed. This table now contains the differentially expressed genes from the conditions we set from the samples of the dataset. The top table resulted in expression of 12,625 genes along with seven variables. Proceeding with the expression, we use dplyr to filter the null values from the DEGs that have been found in the toptables of 3 conditions. After filtration, around we are left with prob ids, which we have to convert into gene symbols/ids for which the library(hgu133plus2.db) is used as it contains mappings between a manufacturer's identifiers and manufacturers accessions. Either you can download the annotation data manually or through a set of R code, which is then read into the R studio environment for mapping purposes. After merging, filtering the required columns and reordering columns, we have successfully produced a genelist for a specific condition i.e., CD vs normal, UC vs normal or combined vs normal. We retrieved around 2,450 entries for condition Ulcerative colitis vs Normal, 5,157 entries for Crohn’s disease vs Normal and 5,072 entries for Combined vs Normal condition.
|
S.no |
gene.symbol |
logfc |
AvgExpr |
t |
P.value |
adj.P.val |
B |
|
1 |
RABGGTA |
0.112717432 |
7.300086 |
1.04211036 |
3.051403e-01 |
0.522784076 |
-5.7643117 |
|
2 |
MAPK3 |
0.011684912 |
8.176038 |
0.06287469 |
9.502559e-01 |
0.976666137 |
-6.2943555 |
|
3 |
TIE1 |
0.316040658 |
3.985100 |
2.71566402 |
1.055796e-02 |
0.082077746 |
-2.9996039 |
|
4 |
CYP2C19 |
0.083767482 |
4.166605 |
0.54176689 |
5.917174e-01 |
0.759267407 |
-6.1507259 |
|
5 |
CXCR5 |
0.200358888 |
5.404175 |
2.21942411 |
3.364000e-02 |
0.150200659 |
-4.0179155 |
Table 3: A demonstration of the example of how a retrieved set of resulted genes with important variable columns such as logFC and P value looks like, the genelist contains upregulated genes responsible for a particular condition in this case Combined UC and CD vs normal.
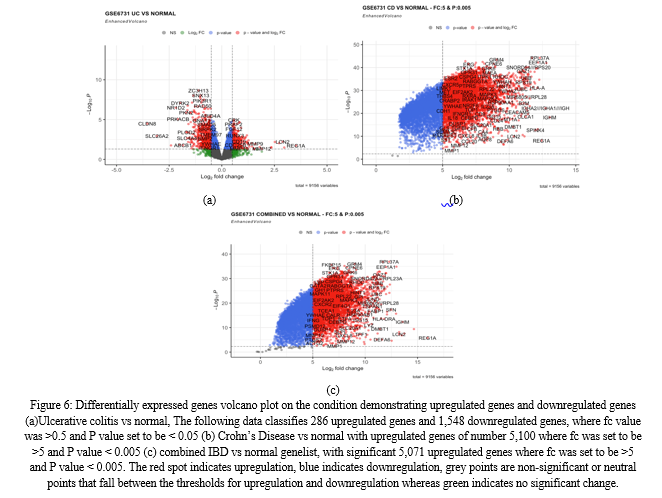
After downloading the gene lists from the R studio environment into the system, visualisation of the genes can be performed using a volcano plot from the function of EnhancedVolcano, which is a useful way to envision differential expressed genes from any specific condition. Depending on the variance of the values of Fold change and P values, we can set a limit which can draw a separation path to investigate the up and down regulated genes.
- Differential Genes Identified for Condition Ulcerative Colitis vs Control
After filtration, a total of 9156 genes were retrieved for this condition, with fold change values ranging from -4 to +4. The statistical significance of p-values varied from 0 to 1. The number of upregulated genes for the condition ulcerative colitis vs control are 286 and 1,548 downregulated genes. Among the top significantly upregulated differentially expressed genes are REG1A, REG3A, LCN2, OLFM4, and MIR8071-2.
The GSE6731 dataset demonstrated that REG1A was apparently higher in the inflamed tissues of active IBD patients relative to healthy tissues. REG1A, being the most significant gene is a protein in the regenerating islet-derived (REG) gene family, is notably highly expressed in inflamed tissues of patients with inflammatory bowel disease (IBD) (Mao; Jia et al., 2021). Within intestinal epithelial cells (IEC), REG1A plays a pivotal role by inhibiting inflammatory responses, promoting cell proliferation, and suppressing epithelial apoptosis. Additionally, other members of the REG gene family, such as REG1B, REG3A, and REG4, which belong to the c-type lectin superfamily, also show significant expression.
On the other side, the most significantly downregulated differentially expressed genes are PLOD2, PKN2, ABCB1, DYRK2, GUCA2A, PRKACB, NR1D2, PCK1, SLC26A2, and CLDN8. CLDN8, or claudin 8, is a critical tight junction (TJ) molecule that is underexpressed in IBD. CLDN8 in the colon prevents absorbed Na+ from leaking into the lumen by sealing the lateral paracellular gap, establishing a protective barrier. It is interesting to note that CLDN8 is the most dramatically downregulated gene in the setting of IBD, indicating a potential role in disease aetiology and intestinal barrier disruption.
PLOD2 is also one of the downregulated genes in UC and according to studies, it is an enzyme that performs post-translational modification on collagen fibrils. Machine learning discovered PLOD2 as a significant gene for identifying Crohn's disease (CD)recurrence after surgery. PLOD2 is a member of the Procollagen-Lysine, 2-Oxoglutarate 5-Dioxygenase Family (PLOD). When PLOD family members are dysregulated, they can cause cancer to develop and metastasis (van Haaften et al., 2020). The differentially expressed genes of IBD for condition Ulcerative colitis are also visualised using the volcano plot by filtering the list according to the log fold change and P values of the respective genes (Figure 6 (a)).
2. Differential genes identified for condition Crohn’s disease vs control
A total of 5,157 genes were retrieved for this condition, with fold change values ranging from 1 to 14. The statistical significance of p-values varied from 0 to 0.045. This condition contains a maximum number of upregulated genes. The number of upregulated genes for the condition Crohn’s disease vs control are of number 5,100. Among the top significantly upregulated differentially expressed genes are IGHM, GAPDH, IGHA1, REG1A, RPL37A, HLA-C, HLA-B, EEF1A1, MIR8071-2, and HLA-A, along with RPL41. Of particular interest, the upregulation of IGHM (Immunoglobulin heavy constant mu) expression has been observed in the context of mucosal inflammation and immune responses associated with inflammatory bowel disease (IBD). Acute exacerbations of chronic IBD, including ulcerative colitis and Crohn's disease, are characterised by an increase in immunoglobulin G (IgG) positive cells in the mucosa (Rüthlein et al., 1992). IGHM - Immunoglobulins are immune system components that can have a role in inflammation. This suggests that IGHM may play a significant role in the immune response and inflammatory processes underlying IBD.
Apart from this, one of the most important genes are HLA-C, HLA-B, HLA-A (the family of Human leukocyte antigen genes) which are most likely the most significant genes related with Crohn's disease. These are major histocompatibility complex (MHC) genes, with certain alleles highly associated with Crohn's disease development and susceptibility. They have an important role in immune response and control, which are critical in the pathophysiology of Crohn's disease and other autoimmune illnesses. The HLA class II genes are thought to play a role in the development of IBD because their products are important in the immune response. Numerous studies have found links between the HLA-DR or -DQ phenotypes and either ulcerative colitis or Crohn's disease (Ahmad et al., 2006). According to the dataset and volcano plot of CD vs Normal ( figure 6(b)), it shows the maximum number of genes are upregulated as compared to the previous gene list.
The downregulation of GCFC2, GABRG2, APOM, CHEK2, and KSR1 in IBD, particularly Crohn's disease, indicates that these proteins may play a role in disease aetiology. These genes play roles in a variety of physiological activities, including transcriptional control (GCFC2), neurotransmission (GABRG2), lipid metabolism (APOM), DNA damage response (CHEK2), and signalling pathways (KSR1).
3. Differential genes identified for condition IBD vs control samples:
This condition yielded 9156 genes with fold change values ranging from -1 to 16. The statistical significance of p-values ranged from zero to 0.05. This condition contains more upregulated genes. The number of upregulated genes for the condition combined IBD samples vs control are of 5,071 upregulated genes whereas downregulated genes were around 3,080 in number. A visual illustration (figure 6 (c)) shows that most of the genes were upregulated for this case as well. One of the top significantly elevated differentially expressed genes in this dataset is REG1A, which is strongly expressed in inflamed tissues of individuals with inflammatory bowel disease (IBD) and plays a role in suppressing inflammatory responses (Mao; Jia et al., 2021).
The group MIR8071-2 and MIR8071-1 contain microRNAs and immunoglobulin heavy chain genes, indicating an aggressive immune response in IBD. OLFM4, which is known for its role in tissue repair and mucosal healing, is also elevated. DEFA5, an antimicrobial peptide vital for host defence, is another notable gene in this dataset.
LCN2, which is involved in innate immunity and increases in response to inflammation, is likewise among the top elevated genes. Furthermore, GAPDH, a popular housekeeping gene frequently employed as a reference in gene expression investigations, had increased expression. The collection of genes IGHA2, IGHA1, IGH, like the previously stated immunoglobulin genes, is elevated, indicating that the immune system plays a role in IBD pathogenesis (Rüthlein et al., 1992). These findings indicate a complex interplay of immune response, inflammation, tissue healing, and antimicrobial defence mechanisms in the context of IBD, emphasising the disease's varied molecular profile (Stallhofer J et al., 2015).
The most significantly downregulated genes for the combined IBD genes were OPRD1, SATB1, RUNX2, SLC16A5 and RAB3GAP1. Among them, SATB1 and RUNX2 have been reportedly found to be least significant in Inflammatory bowel disease. SATB1 is a chromatin remodelling factor required for T cell lineage formation in the thymus. It controls the particular gene expression and activity of effector Th17 cells in tissue inflammation. It affects T-cell growth and immunological responses. SATB1 downregulation has the potential to alter immune cell function and regulation (Yasuda et al., 2019). Another study verified that Runx2 protein levels were shown to be lower in the intestinal epithelial cells (IECs) of CD patients, suggesting that Runx2 may protect IECs against apoptosis in CD, indicating a new biological target for CD treatment. RUNX2 is a transcription factor that plays a role in osteogenesis and bone development, and its downregulation may have an impact on bone remodelling, which has been observed in some IBD patients ( Liugen Gu a 1 et al., 2017).

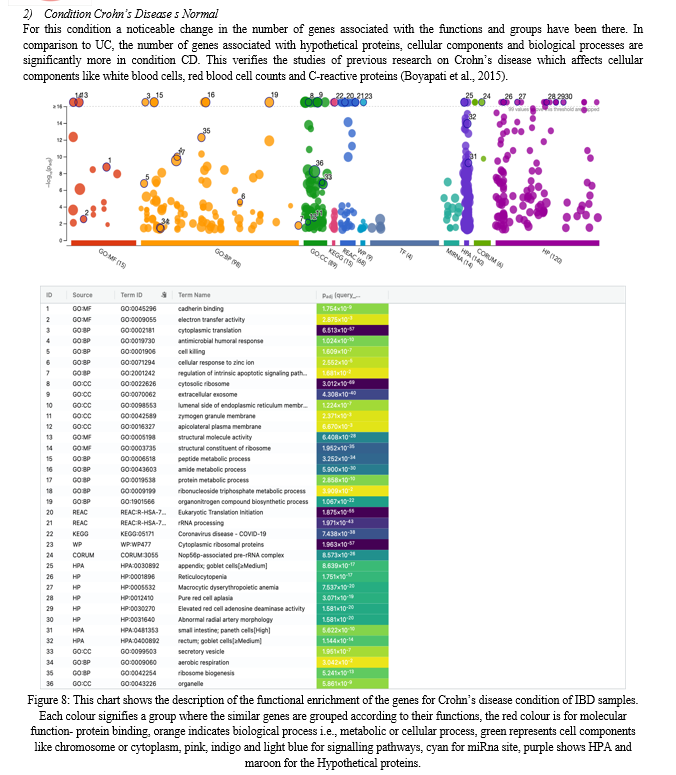

V. DISCUSSION
The prevalence of inflammatory bowel disease (IBD) is on the rise, prompting researchers to seek innovative treatments to alleviate its symptoms and mitigate its impact on patients' lives. IBD, a serious condition with profound consequences, significantly affects patients' overall health and quality of life. While much attention has been devoted to studying IBD in adult patients, there is growing recognition of the importance of understanding its manifestation in children to optimise treatment strategies. Despite considerable efforts in the detection, diagnosis, and management of IBD, uncertainties persist (Seyedian et al., 2019). Previous research has identified a number of genes associated with IBD, providing valuable insights into their role and potential implications for disease progression and comorbidities, including cancer. Notably, innate immunity genes such as NOD2, first identified in 2001, and autophagy genes like ATG16L1 and IRGM underscore the significance of abnormalities in intracellular bacterial processing in the pathogenesis ofInflammatory bowel disease (Noble et al., 2010). Moreover, meta-analyses and subsequent investigations into ulcerative colitis (UC) have pinpointed additional susceptibility genes within the IL-23 pathway, namely IL12B, JAK2, and STAT3, further elucidating the genetic underpinnings of IBD (Ng et al., 2017).
To assess in a conclusive way the role gene in this analysis has been used to derive the characterization of approximately 36 patients with inflamed bowel disease has generated the following conclusions: first, the expression levels of the gene products of HLA class I, REG1A, OLFM4, LCN2, IGHM and DEFA5 differentially associated with progression in patients with IBD. Secondly, the expression levels of these genes have been outlined in the previous research studies. Lastly, these genes are also closely associated with several common events in colon and rectal cancer.
In the present study, the genes REG1A, LCN2, and OLFM4, have been identified as upregulated in ulcerative colitis, each with distinct genetic mechanisms. REG1A, a member of the Reg gene family, has been associated with poor clinical outcomes in early-stage colorectal cancer (CRC) patients (Astrosini et al., 2008). Additionally, REG1A levels were found to be elevated in inflamed colorectal tissues of IBD patients, with abnormal elevations suppressing inflammatory responses, promoting cell proliferation, and reducing epithelial apoptosis in intestinal epithelial cells (IEC) (Derakhshani et al., 2022; Mao et al.). LCN2, also known as neutrophil gelatinase-associated lipocalin (NGAL) or siderocalin, is a peptide created by macrophages, neutrophil granulocytes, and other immunological and parenchymal cells. It exhibits an antibacterial effect by binding catechol-type siderophores with extraordinary affinity. Furthermore, it is associated with a variety of physiological functions, such as transporting hydrophobic ligands across cell membranes, influencing immunological responses, maintaining iron homeostasis, and stimulating epithelial cell differentiation. OLFM4 is a glycoprotein that is naturally expressed in the gastrointestinal system, neutrophils, and the prostate. It participates in innate immunity, inflammation, and cancer. Olfactomedin-4 (OLFM4) is a gene that is up-regulated in IBD tissue and colorectal cancer. It is also present in cells from colon adenocarcinomas. OLFM4 regulates intestinal inflammation and carcinogenesis and may be a therapeutic target for intestinal malignant tumours (Wang et al., 2018).
HLA-A, HLA-B and HLA-C along with IGHM were the prominent genes of Crohn’s disease. The human leukocyte antigen (HLA) complex on chromosome 6p21.3 is the most well-studied genetic locus in inflammatory bowel disease. HLA-A and HLA-B are genes from the human leukocyte antigen (HLA) class I complex, a gene-rich area on chromosome 6p21.3. The conventional HLA genes, HLA-A, HLA-B, and HLA-C, have the strongest association with IBD development. The HLA-A and HLA-B genes may influence the aetiology of IBS by altering the gut immune system. According to one study, the frequency of HLA-B12 was significantly higher in Crohn's disease patients (52%), compared to healthy controls (21%), and ulcerative colitis patients (10%) (Michelakos et al., 2022).
Antimicrobial and cytotoxic peptides known as defensins (DEFA5) play a role in host defence. According to previous studies, aside from their role in the innate immune system and IBD, numerous studies have shown that defensins are expressed and involved in some types of cancer, such as CRC, breast cancer, and kidney cancer. The expression levels of DEFA5 and DEFA6 were considerably higher in CRC tissues than in normal tissues. α-Defensin has five members: DEFA1 (also known as DEFA2, HNP-1, and HNP-2), DEFA3 (also known as HNP-3), DEFA4 (also known as HNP-4), DEFA5, and DEFA6. KLK12, a gene involved in tumour formation, was substantially elevated in CRC among the co-expressed genes DEFA5 and DEFA6 (Arijs et al., 2009).
VI. FUTURE SCOPE AND LIMITATIONS
The future prospects of IBD gene identification through expression profiling are promising, offering significant potential to enhance our comprehension of the molecular mechanisms underpinning inflammatory bowel disease (IBD) and to elevate patient care. One important area of focus is combining multi-omics data, including transcriptomics, proteomics, metabolomics, and genomes, to understand the complex interactions between genetic, epigenetic, and environmental variables that contribute to the pathophysiology of IBD. Researchers can discover new molecular subtypes of IBD, therapeutic targets, and biomarkers by using integrative techniques. This paves the way for personalised medicine approaches that are suited to the unique patient profiles. Furthermore, developments in single-cell sequencing technology offer previously unheard-of clarity in defining gene expression patterns specific to individual cells in the intestinal mucosa, explaining the cellular heterogeneity and immune cell dynamics that drive the pathogenesis of IBD. Furthermore, there is great promise for mining large-scale expression databases and identifying prognostic signatures of disease progression, responsiveness to treatment, and patient outcomes in inflammatory bowel disease (IBD) using artificial intelligence techniques and machine learning algorithms.
To fully realise the potential of IBD gene identification by expression profiling, however, several restrictions and difficulties need to be overcome. One important drawback is that IBD is inherently heterogeneous, with a wide range of clinical presentations, illness histories, and treatment responses. This variability can make it difficult to interpret and repeat gene expression investigations. To enhance robustness and comparability across many research, standardisation of experimental techniques, data analysis pipelines, and quality control measures is essential. Furthermore, the validation of candidate genes and the application of research findings in clinical practice depend critically on the availability of well-characterized patient cohorts with thorough clinical annotations, longitudinal follow-up data, and thorough molecular profiling. In addition, in order to maximise the utility of expression profile data in advancing IBD research and healthcare, ethical questions pertaining to patient privacy, informed permission, and data sharing must be carefully addressed. Despite these obstacles, there are encouraging opportunities to advance IBD gene identification and precision medicine projects through further efforts to combine multidisciplinary skills, make use of cutting-edge technology, and promote collaborative research networks.
The future path of IBD gene discovery using expression profiling entails exploring the dynamic nature of gene expression patterns over time and in response to environmental stimuli, in addition to combining multi-omics data and utilising cutting-edge technologies. Studies that monitor alterations in gene expression patterns across time as the illness develops, progresses, and is treated can provide important information about the molecular processes that underlie IBD flare-ups, remissions, and relapses. In addition, dietary habits, the makeup of the gut microbiota, and host-microbe interactions are important environmental factors that influence gene expression patterns and affect the severity and susceptibility of IBD.
Thus, future studies should aim to clarify the interplay between genetic and environmental factors that contribute to IBD by combining expression profiling with microbiome analysis, dietary evaluation, and environmental exposure data in integrative ways. Furthermore, the development of reliable biomarkers is required for the early identification, precise diagnosis, and prognostic prediction of IBD to translate research findings into clinical practice. To ensure its dependability and efficacy in guiding clinical decision-making, biomarker discovery endeavours should prioritise validation in sizable, well-characterized patient cohorts and assessment of clinical value in real-world situations. Finally, addressing differences in healthcare access, research participation, and resource distribution among various patient groups is critical to advancing inclusive and equitable research in the identification of the IBD gene and enhancing health outcomes for all those afflicted with this intricate and crippling illness.
Aiming to close the gap between fundamental science findings and clinical applications, translational research activities are critical to the advancement of IBD gene discovery by expression profiling. It takes interdisciplinary teamwork between fundamental scientists, doctors, bioinformaticians, and industrial partners to translate research findings into real benefits for patients. It also requires communication with patient advocacy organisations and regulatory bodies. To improve patient outcomes and quality of life, researchers can accelerate the translation of promising biomarkers and therapeutic targets from bench to bedside by cultivating a collaborative environment that promotes information exchange, data sharing, and technology transfer.
Personalised healthcare and precision medicine have the potential to transform the way IBD is managed by customising treatments based on the needs, preferences, and characteristics of each patient. With the use of expression profiling methods, genetic testing, biomarker identification, and computational modelling, physicians can categorise patients into discrete molecular subgroups according to their underlying pathophysiology and anticipated reaction to particular treatments. For patients with IBD, this individualised approach to care maximises therapeutic outcomes, reduces treatment-associated risks and side effects, and enables more focused and effective interventions.
At the same time, efforts are being made to create novel treatments for IBD, with an emphasis on addressing important molecular pathways connected to the aetiology and development of the illness. Novel therapeutic approaches for inflammatory bowel disease (IBD) include gene editing technologies, biologic medicines, small molecule inhibitors, and cell-based therapies. These treatments attempt to control immune responses, restore mucosal homeostasis, and stimulate tissue repair. Through the utilisation of expression profiling, researchers can anticipate treatment responses and find druggable targets, hence accelerating the development and clinical translation of next-generation treatments for inflammatory bowel disease (IBD). This approach will address unmet medical needs and improve patient outcomes globally.
Even with the amazing advancements in IBD research and treatment, there are still a number of problems and unsolved issues. The heterogeneity of IBD, which includes a range of clinical phenotypes, disease trajectories, and treatment outcomes, is a significant obstacle. Personalised medicine methods in IBD require a thorough understanding of the underlying biological mechanisms causing disease heterogeneity as well as the identification of biomarkers that consistently stratify patients into subgroups that are clinically significant. Additionally, in order to guarantee that patients receive the best care possible that is both patient-centered and evidence-based, the long-term safety, efficacy, and cost-effectiveness of developing medicines for IBD must be rigorously evaluated through carefully planned clinical trials and real-world evidence studies. The requirement for improved data sharing and collaboration between researchers, institutions, and stakeholders presents another difficulty for IBD research. Large-scale expression profiling datasets are made easier to aggregate and analyse by open-access data repositories, standardised data formats, and collaborative platforms. This makes it possible for researchers to draw reliable conclusions and validate findings across many cohorts and populations. Data sharing initiatives improve the validity and significance of expression profiling studies in IBD and cultivate a collaborative and innovative culture within the research community by advancing transparency, reproducibility, and scientific rigour.
Looking ahead, IBD gene identification using expression profiling has enormous potential to further our knowledge of the pathophysiology of the disease, improve diagnostic and prognostic instruments, and provide tailored therapeutics that enhance patient outcomes and quality of life. Through the adoption of interdisciplinary approaches, utilisation of state-of-the-art technologies, and emphasis on human centric research, we can expedite the advancement of precision medicine in IBD and establish a more promising future for individuals coping with this intricate and demanding illness.
Conclusion
In conclusion, the study culminated in a thorough investigation into the gene expression profiles associated with inflammatory bowel disease (IBD), with a specific focus on Crohn\'s disease (CD) and ulcerative colitis (UC). Utilising data sourced from the NCBI-GEO database and employing sophisticated analytical methodologies such as pre-processing, normalisation, and differential gene expression analysis, the research team successfully pinpointed key genes implicated in the pathogenesis of IBD. Noteworthy findings included the significant upregulation of genes like REG1A, REG3A, and LCN2 in UC, whereas CD displayed upregulation of IGHM and HLA genes. Furthermore, the comprehensive analysis conducted in this study sheds light on the intricate interplay of genetic factors in the pathogenesis of IBD. Building upon previous research, the identification of upregulated genes in CD, underscores the heterogeneous nature of IBD and highlights distinct molecular signatures associated with each subtype in the gene profiling. These findings not only deepen our understanding of the disease\'s aetiology but also provide potential avenues for personalised therapeutic interventions. Previous research studies had identified HLA and REG3A as particularly influential in IBD development. Moreover, the discovery of downregulated genes, including PLOD2, CLDN8, GCFC2, and GABRG2, offers novel insights into the dysregulated molecular pathways underlying IBD. The differential expression of these genes hints at their crucial roles in modulating immune response, intestinal barrier integrity, and tissue homeostasis, thus implicating them as potential targets for therapeutic interventions aimed at restoring mucosal integrity and ameliorating inflammation. The functional enrichment analysis further underscores the intricate network of biological processes involved in IBD pathogenesis. By elucidating the involvement of identified genes in key pathways related to immune regulation, cytokine signalling, and epithelial barrier function, this study provides a holistic view of the molecular mechanisms driving disease progression. Such insights not only inform our understanding of IBD pathophysiology but also lay the groundwork for the development of targeted therapies tailored to address specific molecular aberrations observed in individual patients. Overall, the findings of this study represent a significant advancement in the field of IBD research, offering valuable insights into the molecular basis of the disease and paving the way for the development of precision medicine approaches aimed at improving patient outcomes and quality of life. Further research aimed at validating these findings in clinical settings and elucidating the functional significance of identified genes is warranted to translate these discoveries into actionable strategies for the diagnosis, prognosis, and treatment of IBD.
References
[1] Jairath, V. and Feagan, B.G. (2020) ‘Global burden of inflammatory bowel disease’, The Lancet Gastroenterology & Hepatology, 5(1), pp. 2–3. doi:10.1016/s2468-1253(19)30358-9. [2] Actis, G.C. et al. (2019) History of inflammatory bowel diseases, Journal of clinical medicine. doi: 10.3390/jcm8111970 [3] Latella, G. (2012) ‘Crucial steps in the natural history of inflammatory bowel disease’, World Journal of Gastroenterology, 18(29), p. 3790. doi:10.3748/wjg.v18.i29.3790. [4] Burisch, J. et al. (2013) ‘The burden of inflammatory bowel disease in Europe’, Journal of Crohn’s and Colitis, 7(4), pp. 322–337. doi:10.1016/j.crohns.2013.01.010. [5] Ahmad, T., Marshall, S.-E. and Jewell, D. (2006) Genetics of inflammatory bowel disease: The role of the HLA complex, World journal of gastroenterology. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4087453/ [6] Cheng, F. et al. (2020) Identification of differential intestinal mucosa transcriptomic biomarkers for ulcerative colitis by bioinformatics analysis, Disease markers. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7596466/ (Accessed: 20 April 2024). [7] de Lange, K.M. et al. (2017) Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease, Nature genetics. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5289481/ [8] Fabian, O. et al. (2023) A current state of proteomics in adult and pediatric inflammatory bowel diseases: A systematic search and Review, International journal of molecular sciences. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10253608/ [9] Federico, A. et al. (2021) ‘Microarray data preprocessing: From experimental design to differential analysis’, Methods in Molecular Biology, pp. 79–100. doi:10.1007/978-1-0716-1839-4_7. [10] Fiocchi, C. (2023) OMICS and multi-omics in IBD: No integration, no breakthroughs, International journal of molecular sciences. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10573814/ (Accessed: 21 April 2024). [11] Frenkel, S. et al. (2019) Genome-wide analysis identifies rare copy number variations associated with inflammatory bowel disease, PloS one. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6559655/ (Accessed: 21 April 2024). [12] Han, L. et al. (2018) Mendelian disease associations reveal novel insights into inflammatory bowel disease, Inflammatory bowel diseases. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6037048/ (Accessed: 21 April 2024). [13] Kedia, S. and Ahuja, V. (2017) Epidemiology of inflammatory bowel disease in India: The great shift east, Inflammatory intestinal diseases. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5988149/#:~:text=The%20overall%20estimated%20IBD%20population,USA%20(with%201.64%20million) (Accessed: 20 April 2024). [14] Kirsner, J.B. (2001) Historical origins of current IBD Concepts, World journal of gastroenterology. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4723519/ (Accessed: 20 April 2024). [15] Mulder, D.J. et al. (2014) A tale of two diseases: The history of inflammatory bowel disease, OUP Academic. Available at: https://academic.oup.com/ecco-jcc/article/8/5/341/616781 (Accessed: 20 April 2024). [16] Rüthlein, J. et al. (1992) Immunoglobulin G (IGG), igg1, and IGG2 determinations from endoscopic biopsy specimens in control, crohn’s disease, and ulcerative colitis subjects., Gut. Available at: https://gut.bmj.com/content/33/4/507 (Accessed: 20 April 2024). [17] Seyedian, S.S., Nokhostin, F. and Malamir, M.D. (2019) A review of the diagnosis, prevention, and treatment methods of inflammatory bowel disease, Journal of medicine and life. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6685307/#:~:text=Ulcerative%20colitis%20is%20associated%20with,commonly%20associated%20with%20rectal%20bleeding. (Accessed: 19 April 2024). [18] Titz, B. et al. (2018) Proteomics and lipidomics in inflammatory bowel disease research: From mechanistic insights to biomarker identification, International journal of molecular sciences. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6163330/ (Accessed: 21 April 2024). [19] Yang, Y.H. et al. (2002) Normalization for cdna microarray data: A robust composite method addressing single and multiple slide systematic variation, Nucleic acids research. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC100354/#:~:text=The%20purpose%20of%20normalization%20is,of%20expression%20levels%20across%20slides. (Accessed: 21 April 2024). [20] Ahmad, T., Marshall, S.-E. and Jewell, D. (2006) Genetics of inflammatory bowel disease: The role of the HLA complex, World journal of gastroenterology. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4087453/ (Accessed: 22 April 2024). [21] Mao; Jia , H. (no date) Protective and anti-inflammatory role of reg1a in inflammatory bowel disease induced by JAK/STAT3 signaling Axis, International immunopharmacology. Available at: https://pubmed.ncbi.nlm.nih.gov/33513463/#:~:text=In%20this%20study%2C%20we%20uncovered,integrity%20of%20intestinal%20mucosal%20barrier. (Accessed: 22 April 2024). [22] van Haaften, W.T. et al. (2020) Intestinal stenosis in crohn’s disease shows a generalized upregulation of genes involved in collagen metabolism and recognition that could serve as novel anti-fibrotic drug targets, Therapeutic advances in gastroenterology. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7457685/#:~:text=Enzymes%20involved%20in%20intracellular%20post%2Dtranslational%20modification%20of,versus%200.40%20%C2%B1%200.38%2C%20p%20=%200.02; (Accessed: 21 April 2024). [23] Stallhofer J;Friedrich M;Konrad-Zerna A;Wetzke M;Lohse P;Glas J;Tillack-Schreiber C;Schnitzler F;Beigel F;Brand S; (no date) Lipocalin-2 is a disease activity marker in inflammatory bowel disease regulated by IL-17A, IL-22, and TNF-? and modulated by IL23R genotype status, Inflammatory bowel diseases. Available at: https://pubmed.ncbi.nlm.nih.gov/26263469/ (Accessed: 22 April 2024). [24] Gu, L. et al. (2018) \'Runt-related transcription factor 2 (RUNX2) inhibits apoptosis of intestinal epithelial cells in Crohn’s disease,\' Pathology, Research and Practice, 214(2), pp. 245–252. https://doi.org/10.1016/j.prp.2017.11.004. [25] Boyapati, R., Satsangi, J. and Ho, G.-T. (2015) Pathogenesis of crohn’s disease, F1000prime reports. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4447044/ (Accessed: 22 April 2024). [26] Assadsangabi, A. et al. (2019) ‘Application of proteomics to inflammatory bowel disease research: Current status and future perspectives’, Gastroenterology Research and Practice, 2019, pp. 1–24. doi:10.1155/2019/1426954. [27] Chan, P.P. (2016) ‘Current application of proteomics in biomarker discovery for inflammatory bowel disease’, World Journal of Gastrointestinal Pathophysiology, 7(1), p. 27. doi:10.4291/wjgp.v7.i1.27. [28] Identification of four potential hematologic biomarkers for the diagnosis of crohn’s disease (2022) Research Square. Available at: https://www.researchsquare.com/article/rs-1612932/v1 (Accessed: 26 April 2024). [29] (29. Kumar, 2019)(2019) Integrating omics for a better understanding of inflammatory bowel disease: A step towards Personalized Medicine - Journal of Translational Medicine, SpringerLink. Available at: https://link.springer.com/article/10.1186/s12967-019-02174-1 (Accessed: 26 April 2024). [30] Latella, G. (2012) ‘Crucial steps in the natural history of inflammatory bowel disease’, World Journal of Gastroenterology, 18(29), p. 3790. doi:10.3748/wjg.v18.i29.3790. [31] Linggi, B. et al. (2021) Meta-analysis of gene expression disease signatures in colonic biopsy tissue from patients with ulcerative colitis, Nature News. Available at: https://www.nature.com/articles/s41598-021-97366-5 (Accessed: 26 April 2024). [32] Loddo, I. and Romano, C. (2015) ‘Inflammatory bowel disease: Genetics, epigenetics, and pathogenesis’, Frontiers in Immunology, 6. doi:10.3389/fimmu.2015.00551. [33] Mahdi, B.M. (2015) ‘Role of HLA typing on crohn’s disease pathogenesis’, Annals of Medicine & Surgery, 4(3), pp. 248–253. doi:10.1016/j.amsu.2015.07.020. [34] Palmieri, O. et al. (2015) ‘Genome-wide pathway analysis using gene expression data of colonic mucosa in patients with inflammatory bowel disease’, Inflammatory Bowel Diseases, p. 1. doi:10.1097/mib.0000000000000370. [35] Yamada, R. et al. (2020) Interpretation of OMICS data analyses, Nature News. Available at: https://www.nature.com/articles/s10038-020-0763-5 (Accessed: 26 April 2024). [36] Singh, D. and Singh, B. (2020) Effective and efficient classification of gastrointestinal lesions: Combining data preprocessing, feature weighting, and improved Ant Lion Optimization - Journal of Ambient Intelligence and humanized computing, SpringerLink. Available at: https://link.springer.com/article/10.1007/s12652-020-02629-0 (Accessed: 26 April 2024). [37] Ahmed, I. et al. (2016) ‘Investigation of faecal volatile organic metabolites as novel diagnostic biomarkers in inflammatory bowel disease’, Alimentary Pharmacology & Therapeutics, 43(5), pp. 596–611. doi:10.1111/apt.13522. [38] Bjerrum, J.T. et al. (2014) Metabonomics of human fecal extracts characterize ulcerative colitis, crohn’s disease and healthy individuals - metabolomics, SpringerLink. Available at: https://link.springer.com/article/10.1007/s11306-014-0677-3 (Accessed: 26 April 2024). [39] Olsen, T. et al. (2016) ‘Normalization of mucosal tumor necrosis factor-?: A new criterion for discontinuing infliximab therapy in ulcerative colitis’, Cytokine, 79, pp. 90–95. doi:10.1016/j.cyto.2015.12.021. [40] Seyedian, S.S., Nokhostin, F. and Malamir, M.D. (2019) A review of the diagnosis, prevention, and treatment methods of inflammatory bowel disease, Journal of medicine and life. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6685307/ (Accessed: 02 May 2024). [41] Ng, S.C. et al. (2017) ‘Worldwide incidence and prevalence of inflammatory bowel disease in the 21st Century: A systematic review of population-based studies’, The Lancet, 390(10114), pp. 2769–2778. doi:10.1016/s0140-6736(17)32448-0. [42] Wang, X.-Y. et al. (2018) ‘Olfactomedin-4 in digestive diseases: A mini-review’, World Journal of Gastroenterology, 24(17), pp. 1881–1887. doi:10.3748/wjg.v24.i17.1881. [43] Arijs, I. et al. (2009) ‘Mucosal gene expression of antimicrobial peptides in inflammatory bowel disease before and after first infliximab treatment’, PLoS ONE, 4(11). doi:10.1371/journal.pone.0007984. *******************
Copyright
Copyright © 2024 Payal Priyadarshini, Ankur Chaurasia. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET62654
Publish Date : 2024-05-24
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online